- Автоматизация
- Антропология
- Археология
- Архитектура
- Биология
- Ботаника
- Бухгалтерия
- Военная наука
- Генетика
- География
- Геология
- Демография
- Деревообработка
- Журналистика
- Зоология
- Изобретательство
- Информатика
- Искусство
- История
- Кинематография
- Компьютеризация
- Косметика
- Кулинария
- Культура
- Лексикология
- Лингвистика
- Литература
- Логика
- Маркетинг
- Математика
- Материаловедение
- Медицина
- Менеджмент
- Металлургия
- Метрология
- Механика
- Музыка
- Науковедение
- Образование
- Охрана Труда
- Педагогика
- Полиграфия
- Политология
- Право
- Предпринимательство
- Приборостроение
- Программирование
- Производство
- Промышленность
- Психология
- Радиосвязь
- Религия
- Риторика
- Социология
- Спорт
- Стандартизация
- Статистика
- Строительство
- Технологии
- Торговля
- Транспорт
- Фармакология
- Физика
- Физиология
- Философия
- Финансы
- Химия
- Хозяйство
- Черчение
- Экология
- Экономика
- Электроника
- Электротехника
- Энергетика
Лабораторна робота №4-5 3 страница
Таблиця 3. 1. Парціальні термодинамічні характеристики води (в кал/моль) в деяких набряклих високомолекулярних речовинах
| Висо-комоле-кулярна речовина | t, °С | Δ Н1 | Δ G1 | TΔ S1 | Δ H1 | Δ G1 | TΔ S1 |
| (5, 7% Н2О, 0, 943 мол. дол. речовини) | (10, 8% Н2О, 0, 892 мол. дол. речовини) | ||||||
| Агар | -3200 | -1850 | -1350 | -1850 | -1000 | -850 | |
| Казеїн | -2050 | -900 | -1150 | -1000 | -350 | -650 | |
| Кератин | -2250 | -1200 | -1050 | -1150 | -500 | -950 | |
| » | -2800 | -1100 | -1400 | -1650 | -450 | -1250 | |
| Целюлоза | -1000 | -230 | -770 | -200 | -30 | -175 | |
3. 4. Стадії набухання та розчинення полімерів
Набухання полімеру в рідині характеризується мірою набухання (α ), що розраховується за рівнянням:
α =(m-m0)/ m0 (3. 1)
де m0 та m - наважка полімеру до і після набухання.
Міра набухання чисельно дорівнює масі (в г) рідини, поглиненою 1 г високомолекулярної речовини. При визначенні α слід враховувати, що в ході набухання з полімеру вимиваються низькомолекулярні фракції, і тому обчислена за наведеною вище формулою міра набухання може бути декілька нижча за ту. яка відповідала б набуханню в парах розчинника.
Визначаючи міру набухання через задані проміжки часу, можна отримати криві, що характеризують кінетику набухання. Часто про набухання судять не за приростом маси, а за збільшенням об'єму зразку. На рис. 3. 1 зображені типові кінетичні криві набухання.
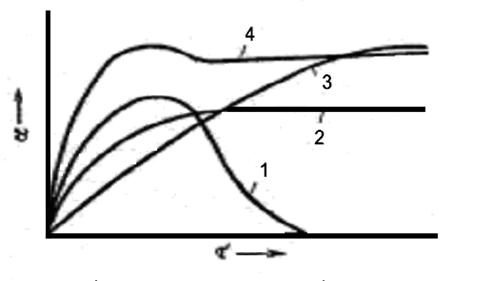
Рис. 3. 1. Типи кінетичних кривих набрякання: 1- неорганічне набрякання; 2 – швидке набрякання з малим значенням граничного набрякання; 3 – швидке набрякання з великим значенням граничного набрякання; 4 – обмежене набрякання з екстрагованою низькомолекулярною фракцією.
Криві 3 і 4 характеризують обмежене набухання, при цьому крива 3 відповідає полімеру, що швидко набухає, але з малим значенням граничного набухання, а крива 4 - полімеру, що повільно набухає, з великим значенням граничного набухання. Як видно, обидві криві спочатку круто підіймаються, а потім плавно переходять в пряму, паралельну осі абсцис. Аналітично ці криві можна представити диференційним рівнянням:
dα /dτ = k(α макс-α τ ) (3. 2)
де α макс - міра граничного набухання; α τ - міра набухання на момент часу τ; k - константа швидкості набухання, яка залежить від природи полімеру, природи розчинника та температури.
В результаті інтегрування рівняння приймає вигляд:
k = (1\τ ) ln α макс/( α макс-α τ ) (3. 3)
Швидкість набухання різних полімерів треба порівнювати за нахилом дотичних, які проведені до кривих від початку координат, а здатність полімерів до набухання слід характеризувати за граничною мірою набухання.
Крива 1 (рис. 3. 1) характеризує необмежене набухання, коли полімер розчиняється в розчиннику. В цьому випадку про граничну міру набухання говорити не можна, хоча на кривій і є максимум. Крива 4 характеризує обмежене набухання, коли з речовини, що набухає, екстрагується значна кількість низькомолекулярних фракцій, що зменшує граничну міру набухання.
На набухання в значній мірі може впливати форма зразку. Тому досліди, в яких хочуть порівняти набухання різних полімерів, потрібно проводити завжди із зразками однакової форми.
Набухання високомолекулярної речовини може призвести до виникнення значного тиску, якщо щось заважає збільшенню об'єму зразку.
Наприклад, при набуханні деревини у воді може розвиватися тиск в десятки атмосфер. Цей тиск настільки великий, що в давні часи це явище використовували для подрібнення скелі: в щілини скелі забивали дерев’яні клини і потім заливали воду, клини набухали у воді, прагнучи збільшити свої розміри і тим самим створювали тиск, який руйнував скелю.
Тиск (р), який розвивається при набуханні, можна обчислити за рівнянням Пізняка:
p = p0 ck (3. 4)
де р0 - константа, яка залежить від природи високомолекулярної речовини, розчинника і температури; с - вміст сухого полімеру у драглі, що набрякає; k - константа, значення якої зазвичай близька до 3. Вочевидь, що при с=1 значення р повинно дорівнювати р0. При логарифмуванні рівняння Пізняка приймає вигляд:
ln p = ln p0 + k ln c (3. 5)
Це рівняння прямої дозволяє графічно легко визначити величини р0 і k, а при відомих константах і при заданому значенні с можна визначити і р.
Для прикладу на рис. 3. 2 представлена логарифмічна залежність тиску від концентрації для каучуку, що набухає в різних розчинниках. 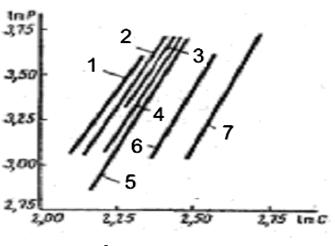
Рис. 3. 2. Залежність ln p від ln c при набряканні натурального каучуку в різних розчинниках: 1 – чотирьох хлористий вуглець; 2 – хлороформ; 3 – тетрахлоретан; 4 – толуол; 5 – бензол; 6 – діетиловий ефір; 7 – дихлоретан.
Для всіх випробуваних розчинників залежність між ln p і ln c має лінійний характер і константа не змінюється при переході від однієї рідини до іншої. Цікаво, що якщо рідини, в яких каучук, що набрякав, розташувати в порядку збільшення константи р0, то вони утворюють ряд, який співпадає з рядом, в якому ці ж рідини стоять в порядку збільшення граничної міри набухання в них полімеру. Це показує зв'язок між тиском набухання та граничною мірою набухання.
Окрім збільшення об'єму високомолекулярної речовини, на початку набухання часто спостерігається зменшення об'єму всієї системи і тепловий ефект набухання. Ці явища дуже істотні для розуміння механізму набухання.
В той час, як при набуханні об’єм полімеру завжди збільшується, об'єм всієї системи (полімер + розчинник) зазвичай зменшується.
Це особливо помітно при набуханні полярних полімерів в полярних розчинниках. Зменшення об'єму системи при набуханні, яке називається контракцією, в більшості випадків добре описується наступним емпіричним рівнянням зa двома константами:
V = α m/(β + m) (3. 6)
де V – концентрація; m - маса рідини, яка поглинена при набуханні 1 г полімеру; α та β - константи.
Контракція системи при набуханні полімеру пояснюється орієнтацією молекул розчинника в результаті їх «адсорбції» макромолекулами, що сприяють збільшенню щільності системи. Крім того, частково контракція відбувається за рахунок чисто стеричного чинника - при набуханні малі молекули розчинника проникають в простір між великими макромолекулами, внаслідок чого компактність упаковки молекул збільшується.
Тепловий ефект при набуханні високомолекулярної речовини зазвичай позитивний, наприклад, при набуханні полярних полімерів в полярних же розчинниках. Це і зрозуміло, оскільки виділення тепла при набуханні пов'язане зі взаємодією молекул полімеру з розчинником. Розрізняють інтегральну теплоту набухання qінт, тобто загальну кількість тепла, що виділилося при набуханні 1 г сухого полімеру, та диференціальну теплоту набухання qдиф, що є кількістю тепла, яке виділилося при поглинанні 1 г рідини сухою або вже набряклою високомолекулярною речовиною.
В якості прикладу в таблиці 3. 2 приведені інтегральні та диференціальні теплоти набухання деяких високомолекулярних речовин (за даними Каца та Марка).
Таблиця 3. 2. Інтегральні і диференціальні теплоти набухання (в кал/г) деяких високомолекулярних речовин
| Високомолекулярна речовина | Розчинник | qінт | qдиф (для сухої речовини) |
| Казеїн | Вода | 25, 4 | |
| Нуклеїн | » | 23, 4 | |
| Індулін | » | 21, 8 | |
| Дерев’яне волокно | » | 16, 9 | - |
| Целюлоза | » | 10, 7 | |
| Ацетат целюлози | Трихлоретилен | 11, 4 | |
| Теж саме | Бензилхлорид | 8, 1 | |
| » | Бензиловий спирт | 8, 2 | |
| Нітрат целюлози | Мурашина кислота | 6, 8 | |
| Теж саме | Етиловий спирт | 5, 5 |
Інтегральна теплота набухання qінт складається з трьох величин:
qінт' — теплота, яка відповідає роботі, необхідній для роз’єднання макромолекул, qінт'' - теплота, яка відповідає роботі роз’єднання молекул розчинника, та qінт''' - теплота, що виділяється в результаті взаємодії розчинника з високомолекулярною речовиною. Перші дві величини мають завжди негативне значення, третя -позитивне:
qінт = -qінт' - qінт''+ qінт ''' (3. 7)
При набуханні неполярної високомолекулярної речовини в неполярній рідині абсолютні значення величин qінт' , qінт '', qінт ''' близькі та незначні, тому тепловий ефект близький до нуля. При набуханні полярної високомолекулярної речовини в полярному розчиннику величина qінт''', як правило, максимальна і розчинення проходить з виділенням тепла. Для кристалічних високомолекулярних речовин величина qінт' зазвичай висока. У цьому випадку теплота набухання може бути негативною, внаслідок чого набухання проходить важко.
Диференціальна теплота набухання тим менша, чим більше міра набухання високомолекулярної речовини. В якості прикладу приведемо знайдені інтерполяцією диференціальні теплоти набухання для желатину (табл. 3. 3):
Таблиця 3. 3. Диференціальні теплоти набухання для желатину
| Маса води, поглинена | |||||
| 1 г желатину, г | 0, 000 | 0, 005 | 0, 041 | 0, 103 | 0, 242 |
| qдиф, кал/г |
Як можна бачити, диференціальна теплота набухання швидко падає зі збільшенням гідратації високомолекулярної речовини.
Інтегральна теплота набухання qінт може бути представлена рівнянням, аналогічним рівнянню контракції:
qінт = ai/(b+i) (3. 8)
де і - міра гідратації, а і b - константи.
Істотно. що відношення контракції до інтегральної теплоти набухання є, як правило, величиною постійною, тобто
V/ qінт =const (3. 9)
Це вказує на те, що контракція та виділення тепла при набуханні зазвичай є процесами взаємно зв'язаними, які в основному протікають в результаті взаємодії молекул високомолекулярної речовини з дифундуючими в неї молекулами розчинника, тобто в результаті сольватації (гідратації).
Звертає на себе увагу, що рівняння (3. 6) схоже з рівнянням адсорбції Ленгмюра. Це є ще одним доказом того, що сольватація має характер адсорбційного явища.
Наприкінці розглянемо вплив температури на обмежене набухання. Вплив температури на набухання легко визначити виходячи з термодинамічного розглядання процесу. Якщо набухання є екзотермічний процес, що характерно для першої стадії набухання, то рівноважна міра набухання знижується з підвищенням температури. Відповідно до цього, обмежене набухання, наприклад целюлози у воді або в розчині їдкого натрію, представляє типовий екзотермічний процес, який сильно гальмується з підвищенням температури. Проте, як ми бачили, на другій стадії набухання може бути ендотермічним процесом. В цьому випадку набухання повинне збільшуватися із зростанням температури. Досліди показали, що набухання желатину загалом з підвищенням температури збільшується. Швидкість набухання з підвищенням температури, звичайно, завжди повинна зростати, оскільки підвищення температури при всіх обставинах сприяє прискоренню встановлення рівноважного стану системи.
Тема 4. Термодинаміка розчинення
високомолекулярних речовин
План:
4. 1. Термодинамічна характеристика процесу розчинення ВМС.
4. 2. Вплив природи розчинника і полімеру на значення термодинамічних показників процесу розчинення.
4. 1. Термодинамічна характеристика процесу розчинення ВМС.
Розчинення високомолекулярних речовин прийнято розглядати як процес змішання двох рідин. Ця точка зору приймає до уваги як енергетичну взаємодію між молекулами речовини, що розчиняється, і розчинника, так і дію ентропійного чинника, який зумовлює рівномірне розподілення молекул розчиненої речовини в розчині. Аналогія між розчиненням високомолекулярної речовини і процесом змішанням двох рідин не є формальною, а відповідає самій суті явища. Так, обмежене набухання високомолекулярної речовини відповідає процесу обмеженого змішання, а необмежене набухання, що переходить в розчинення, — процесу необмеженого змішання.
Самовільне розчинення високомолекулярної речовин, як і будь якої іншої речовини, при постійному тиску повинно супроводжуватися зменшенням ізобарно-ізотермічного потенціалу (вільної енергії при постійному тиску).
Згідно другому закону термодинаміки зміна ізобарно-ізотермічного потенціалу системи складає:
Δ G=Δ H –TΔ S (4. 1)
де H – ентальпія; Т – абсолютна температура; S – ентропія.
Коли система не змінює свого об'єму, що з певним припущенням можна віднести до процесу розчинення, рівняння (4. 1) можна замінити наступним:
Δ F=Δ U – TΔ S (4. 2)
де F – ізохорно-ізотермічний потенціал (вільна енергія); U – внутрішня енергія.
Вочевидь, для того, щоб сталося самовільне розчинення полімеру, Δ G або Δ F повинні мати негативне значення. Це може бути у двох випадках.
1. За умови Δ H< 0 (або Δ U< 0), яка дотримується, якщо при розчиненні виділяється тепло, оскільки зміна ентальпії (або внутрішньої енергії) дорівнює інтегральній теплоті розчинення із зворотним знаком. Така умова часто виконується на практиці, наприклад, при розчиненні полярних полімерів у полярних розчинниках. Позитивний тепловий ефект при розчиненні пояснюється тим, що теплота сольватації макромолекул більше теплоти власне розчинення, а, як відомо, загальний тепловий ефект розчинення дорівнює алгебраїчній сумі теплот сольватації і власне розчинення.
2. За умови Δ S> 0, яка завжди виконується на практиці при розчиненні, оскільки ентропія змішання завжди додатна.
Як відомо, молярна ентропія змішання двох речовин для ідеальної системи або зміна ентропії системи при розчиненні деякої кількості речовини в даному об'ємі розчинника, знаходиться за наступним термодинамічним рівнянням:
Δ Sід= –R(n1lnN1 + n2lnN2) (4. 3)
де R – універсальна газова стала; n1 і n2 – число молей відповідно розчинника та полімеру, що змішують; N1 і N2 – мольні долі обох компонентів.
Парціальні ж молярні ентропії змішання можна обчислити за рівняннями:
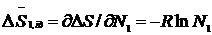 (4. 4)
(4. 4)
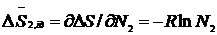 (4. 5)
(4. 5)
При обчисленні ідеальної ентропії змішання використовують уявлення про ізодіаметричні молекули. Тоді підвищення молекулярної маси одного компонента повинно призводити до зменшення числа молекул цього компонента в 1 г системи і, відповідно, до зменшення ентропії змішання даних вагових кількостей обох речовин. Ентальпія (або внутрішня енергія) не повинна змінюватися, оскільки вагові кількості компонентів, що змішуються, а отже, і число атомних груп, що взаємодіють, не змінюються. Звідси витікає, що по мірі підвищення молекулярної маси полімеру значення члена TΔ S в рівняннях (4. 1) і (4. 4) повинно зменшуватися і відчутно зростати значення члену Δ H (або Δ U). Проте при дослідженнях розчинення високомолекулярних речовин з ланцюговими молекулами було встановлено, що подібна залежність дотримується в тій чи іншій мірі лише для полімерів з жорсткими молекулами. При розчиненні ж гнучких ланцюгових молекул в низькомолекулярних рідинах, як показав ряд досліджень, ентропія змішання зазвичай значно перевищує ентропію змішання ідеальної системи, причому тим більше, чим більше молекулярна маса полімеру. Аномально велика ентропія змішання при розчиненні полімерів, яка перевищує в сотні і тисячі разів ідеальну, пояснюється тим, що при змішанні сильно зростає можливість руху в розчині окремих ділянок (сегментів) гнучких ланцюгових молекул. Можна вважати, що в розчині сегменти макромолекул відіграють, по суті, роль окремих кінетичних одиниць.
Згідно статистичній фізиці збільшення ентропії при розчиненні високомолекулярних речовин пояснюється тим, що макромолекули в розчині можуть бути розташовані різним чином, причому кожна макромолекула може мати велике число конформацій. У не розчиненому полімері ланцюги молекул заважають один одному приймати довільні конформації. За наявності розчинника взаємний вплив молекулярних ланцюгів стає все меншим і, нарешті, в гранично розбавленому розчині, коли макромолекули дуже далеко одна від одної, вони можуть приймати практично будь-які конформації. Інакше кажучи, термодинамічна вірогідність стану макромолекул в малов'язкому розчині більше, ніж у вихідному полімері.
Позначимо через Wн початкову термодинамічну вірогідність стану макромолекул (до розчинення) і через Wк – кінцеву (після розчинення). Як відомо, вірогідність W пов’язана з ентропією S рівнянням Больцмана:
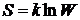 (4. 6)
(4. 6)
де k—постійна Больцмана.
Тоді зміна ентропії під час переходу речовини в розчин буде:
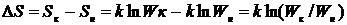 (4. 7)
(4. 7)
Оскільки вірогідність Wк завжди більше Wн то приріст ентропії розчинення завжди позитивний. З приведеного рівняння видно, що зі збільшенням вірогідності в результаті розчинення полімеру ентропія системи завжди зростає.
Зі сказаного можна зробити важливий висновок, що високомолекулярні речовини з гнучкими макромолекулами повинні завжди краще розчинятися, ніж з жорсткими, оскільки перші можуть розташовуватися в розчині значно більшим числом способів. Крім того, слід пам'ятати, що у жорстких макромолекул, зазвичай орієнтованих більш менш паралельно, енергія взаємодії між окремими молекулярними ланцюгами дуже велика, і такі ланцюги важко відірвати один від одного. Цими обставинами і можна пояснити зазвичай вельми обмежене число розчинників для високомолекулярних речовин з жорсткими ланцюгами (целюлоза, полівінілхлорид, поліаміди).
Як вже було вказано, для розчинення суттєвим є дотримання однієї умови – зменшення ізобарно-ізотермічного потенціалу системи. Тому розчинення може здійснюватися і тоді, коли тепловий ефект негативний, але ентропія системи при розчиненні зростає настільки, що в кінці кінців обумовлює зменшення ізобарно-ізотермічного потенціалу.
При підвищенні температури значення чинника ентропії стає все більшим, і, таким чином, для будь-якої високомолекулярної речовини і розчинника повинна існувати критична температура розчинення Ткрит, вище якої спостерігається їх змішування в усіх співвідношеннях. Теоретично така критична температура повинна існувати для будь-якої комбінації високомолекулярної речовини і розчинника. Практично ж вона у багатьох випадках не може бути досягнута через низьку температуру кипіння розчинника і низьку температуру деструкції високомолекулярної речовини. Критичну температуру змішання легко знайти за умови
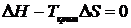 , (4. 8 а), тоді
, (4. 8 а), тоді 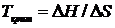 , (4. 8 б)
, (4. 8 б)
4. 2. Вплив природи розчинника і полімеру на значення термодинамічних показників процесу розчинення
Досвід останніх років показав, що при розчиненні полімерів чинник ентропії завдяки аномально великому значенню ентропії змішання в деяких випадках грає основну роль. Наприклад, він майже цілком визначає розчинність неполярних полімерів у вуглеводнях. Тому виявилися неправими ті дослідники (Кац, Бренстед), які намагалися пояснити розчинність полімерів лише впливом ентальпійного чинника. Проте, коли мають справу з полярними полімерами, наприклад, при розчинені желатину у воді, або нітрату целюлози в ацетоні, ентропійний чинник може грати істотну роль і його впливом не можна нехтувати.
Розглянемо причини, що обумовлюють розчинення полімерів, на деяких типових прикладах. Для цього скористаємося таблицею 4. 1.
Як видно з даних, приведених в таблиці 4. 1, розчинення полярного нітрату целюлози в полярному циклогексаноні йде як внаслідок зменшення ентальпії ( 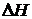 < 0), так і в результаті збільшення ентропії (
< 0), так і в результаті збільшення ентропії ( 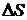 > 0).
> 0).
Таблиця 4. 1. Співвідношення між термодинамічними величинами при розчиненні деяких високомолекулярних сполук
| Система | Характер розчинення |
|
| 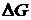
|
| Нітрат целюлози в циклогексаноні | Екзотермічний | < 0 | ³ 0 | < 0 |
| Яєчний альбумін у воді | » | < 0 | < 0 | < 0 (при 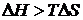 ) )
|
| Полі-ізо-бутилен в ізо-октані | Ізотермічний | > 0 | < 0 | |
| Каучук в толуолі | Ендотермічний | > 0 | > 0 | < 0 (при 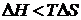 ) )
|
Завдяки тому, що змішанню сприяють обидва чинника, циклогексанон є хорошим розчинником нітрату целюлози.
При розчиненні полярного яєчного альбуміну у воді (полярний розчинник) процес спочатку йде лише за рахунок зменшення ентальпії. Ентропія на початковій стадії розчинення має навіть негативні значення через сильну гідратацію полімеру. Молекули води в оболонці гідрату переходять в більш впорядкований стан. Крім того, не виключено зменшення гнучкості молекул полімеру при гідратації його молекул. Але, зазвичай, для того, щоб полімер міг розчинятися, абсолютне значення має бути більше 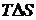 . Проте по мірі зростання гідратації молекул полімеру, вони розчиняються у надлишку води зі все меншим виділенням тепла. Розчинення на цій кінцевій стадії йде вже головним чином, за рахунок можливості розподілу молекул у всьому об’ємі системи, тобто вже за рахунок чинника ентропії.
. Проте по мірі зростання гідратації молекул полімеру, вони розчиняються у надлишку води зі все меншим виділенням тепла. Розчинення на цій кінцевій стадії йде вже головним чином, за рахунок можливості розподілу молекул у всьому об’ємі системи, тобто вже за рахунок чинника ентропії.
Розчинення неполярного полі-ізо-бутилену в неполярному ізо-октані йде лише внаслідок підвищення ентропії без виділення тепла, тобто змішання має тут ізотермічний характер. Істотно, що при розчиненні полі-ізо-бутену в ізо-октані, гідратованому димері ізо-бутилені, енергетичний бар’єр при обертанні окремих ланок ланцюгу молекули не змінюється, так як дія міжмолекулярних сил в розчині така ж, що і в самому полі-ізо-бутилені. Іншими словами, розчинення в цьому випадку відбувається без зміни гнучкості макромолекул.
Нарешті, розчинення каучуку в толуолі йде теж за рахунок збільшення ентропії. Відмінність цього випадку від попереднього полягає у тому, що тут спостерігається невеликий негативний тепловий ефект. Проте цей ефект із надлишком перекривається збільшенням ентропії, оскільки молекули каучуку вельми гнучкі.
Представлені в таблиці 4. 1 комбінації зміни ентальпії і ентропії повністю охоплюють усі типові випадки розчинення полімерів. При інших комбінаціях цих величин розчинення не відбувається, оскільки в таких випадках > 0.
Тема 5. Дублення
План:
5. 1. Дублення.
5. 2. Вулканізація.
5. 3. Деструкція полімерів.
5. 4. Механічна деструкція.
5. 5. Термічна деструкція.
5. 6. Фотохімічна деструкція.
5. 7. Ультразвукова деструкція.
5. 1. Дублення
Дублення – поширений різновид структурування біополімерів. Воно представляє цікавість не тільки при структуруванні білків у виробництві натуральної шкіри, але і при виготовленні білкових клеїв, шкір, картонів та ін. Дублення, напевне, буде в подальшому відігравати важливу роль у виробництві нових видів штучної шкіри.
Певні речовини при взаємодії з білками проявляють властивості, які спричиняють дублення білків. До них відносяться неорганічні сполуки: солі хрому, алюмінію, заліза, кремнію, титану, цирконію та ін., органічні сполуки рослинного походження (екстракти), жири, масла, альдегіди, а також синтетичні продукти (синтани) типу поліфенолів, сульфокислот і деякі промислові відходи (лігносульфонові кислоти).
В технології одержання матеріалів для виготовлення взуття використовуються солі хрома, алюмінію, рослинні екстракти, жири та альдегіди. В результаті недостатнього вивчення таких складних поліфункціональних об’єктів, як білкові речовини, на сьогодні немає однозначних уявлень про механізму дублення. Однак, в цілому ефект дублення безсумнівно є наслідок змішаного структурування білків сполуками, які викликають дублення з утворенням широкого набору просторових хімічних, ковалентних, координаційних зв’язків та всіх видів міжмолекулярної взаємодії.
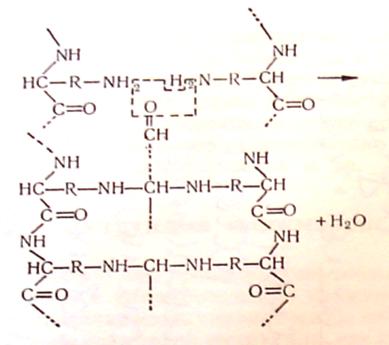
Так, при дубленні білків катіонними комплексними сполуками хрому утворюються поперечні зв’язки за рахунок включення кислотних (-COOH) та основних (-NH2) груп бокових частин білкових ланцюгів у внутрішню сферу хромових комплексів.
Для аніонних комплексів аналогічна взаємодія, проходить, ймовірніше, за участю основних груп. Одночасно з координаційними зв’язками в цьому випадку можливо утворення інших типів зв’язків.
Одна з найпростіших схем структурування при хромовому дубленні має наступний вигляд:
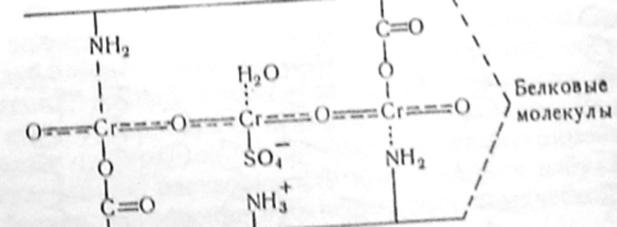
Схема 5. 1. Структурування білків при хромовому дубленні.
Дія інших дубильних мінеральних солей в цілому анологічна.
При дубленні жирами, які містять залишки легкоокислюваних ненасичених жирних кислот, не виключається утворення наступних видів поперечних зв’язків:
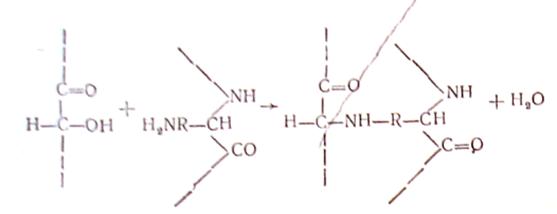
Схема 5. 2. 1. Структурування білків при дубленні жирами.
 .
.
Схема 5. 2. 2. Структурування білків при дубленні жирами.
В цьому випадку активною сполукою є перекис. Дублення прискорюється при збільшенні притоку кисню повітря, при помірному підвищенні температури, присутності каталізаторів (сиккативів) та ін.
Рослинні екстракти, як складні поліфункціональні сполуки типу депсидів, поліфенолокислот та ін., імовірно, можуть структурувати білки за рахунок різних видів зв’язку, які умовно можна описати наступною схемою:
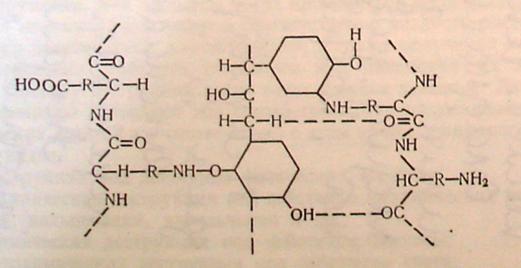
Схема 5. 3. Структурування білків при дубленні рослинними екстрактами.
Дубильну дію альдегідів, внаслідок відносної простоти їх будови, вивчено більш повно. Так, вже не викликає сумнівів той факт, що при взаємодії білка з альдегідом останній переважно реагує з основними (-NH2) групами білкових ланцюгів, блокуючи їх:
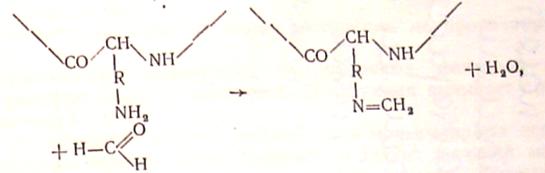
Або «зшиває» групи сусідніх ланцюгів,
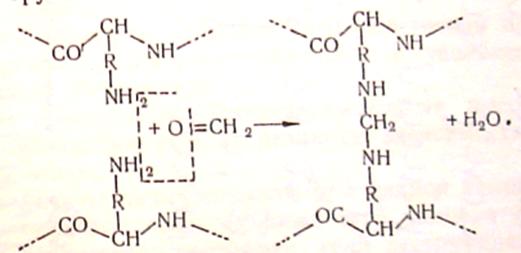
Схема 5. 4. Структурування білків при дубленні альдегідами.
|
|
|
© helpiks.su При использовании или копировании материалов прямая ссылка на сайт обязательна.
|