- Автоматизация
- Антропология
- Археология
- Архитектура
- Биология
- Ботаника
- Бухгалтерия
- Военная наука
- Генетика
- География
- Геология
- Демография
- Деревообработка
- Журналистика
- Зоология
- Изобретательство
- Информатика
- Искусство
- История
- Кинематография
- Компьютеризация
- Косметика
- Кулинария
- Культура
- Лексикология
- Лингвистика
- Литература
- Логика
- Маркетинг
- Математика
- Материаловедение
- Медицина
- Менеджмент
- Металлургия
- Метрология
- Механика
- Музыка
- Науковедение
- Образование
- Охрана Труда
- Педагогика
- Полиграфия
- Политология
- Право
- Предпринимательство
- Приборостроение
- Программирование
- Производство
- Промышленность
- Психология
- Радиосвязь
- Религия
- Риторика
- Социология
- Спорт
- Стандартизация
- Статистика
- Строительство
- Технологии
- Торговля
- Транспорт
- Фармакология
- Физика
- Физиология
- Философия
- Финансы
- Химия
- Хозяйство
- Черчение
- Экология
- Экономика
- Электроника
- Электротехника
- Энергетика
3.Терапия второй линии.
Вторая линия терапии применяется при отсутствии ответа на лечение, рефрактерном течении заболевания или его прогрессировании, несмотря на проведенное лечение первой линии. Выбор препарата зависит от возраста пациента, степени тяжести поражения крови, общего соматического статуса и предыдущих видов лечения.
В терапии второй линии при лечении СС используются следующие препараты:
1. Хлорамбуцил (Лейкеран) в сочетании с системными глюкокортикостероидами: хлорамбуцил 2-12 мг/день + преднизолон 20 мг/день. Основным побочным эффектом является лейкопения, к ранним побочным эффектам относятся миело- и иммуносупрессия, гиперурикемия, к отсроченным – аменорея, бесплодие, интерстициальный фиброз легких, цистит, гепатотоксичность, периферическая нейропатия.
2. Пегилированный липосомальный доксорубицин: вводится в дозе 20-30
мг/м2 в/в каждые 2-4 недели.
3. Ингибиторы гистондеацетилаз (HDACi) (вориностат). Назначается перорально по 400 мг ежедневно до достижения полного контроля (отсутствие признаков дальнейшего прогрессирования) или же до появления признаков неприемлемой токсичности. Из побочных эффектов встречаются тромбоцитопения, анемия, анорексия, тошнота, мышечные спазмы.
4. Гемцитабин (Гемзар): 1200 мг/м2 в 1, 8 и 15 день 28-дневного цикла (3-6 курсов). Препарат хорошо переносится, из побочных эффектов наблюдаются нейтропения, тромбоцитопения и анемия.
5. Деоксикоформицин (Пентостатин): 4-8 мг/м2/день 3 дня каждые 28 дней. Побочные эффекты: гематологические, гастроинтестинальные.
6. Сочетание флударабина (25 мг/м2 каждые 3-4 недели) и циклофосфамида
(250 мг/м2/день 3 дня 1 раз в месяц) в течение 3-6 месяцев.
7. Аллогенная трансплантация костного мозга может рассматриваться как потенциально возможный вид лечения у пациентов с СС с агрессивным течением и отсутствием эффекта от стандартных режимов терапии.
4. Поддерживающая терапия
Наружные и системные (10-20 мг преднизолона/сут) глюкокортикостероиды используются в виде поддерживающей терапии у пациентов с СС. При длительном применении их отмена обычно ассоциирована с рецидивом заболевания, побочные эффекты включают атрофию кожи (при длительном наружном применении) и подавление
функции надпочечников и/или остеопороз (при распространенной аппликации наружных или длительном приеме системных глюкокортикостероидов).
К дополнительным видам терапии относится фототерапия: ПУВА- терапия и узковолновое УФО спектра В (311 нм) (см. главу «Грибовидный микоз»).
Применение лейкафереза улучшает результаты стандартных видов терапии, уменьшает зуд и количество клеток Сезари в крови.
Тотальное облучение кожи (ТОК) в дозе 20-40 Гр рекомендовано комбинировать с другими видами системной терапии или применять как монотерапию с паллиативными целями.
Большое значение в ведении пациентов с СС имеет терапия, направленная на снижение интенсивности зуда и различных нейропатий (ощущений жжения, боли, стягивания кожи, парестезий). Для уменьшения этих ощущений используются увлажняющие кремы и антигистаминные препараты. Известно, что кожа больных СС избыточно колонизирована S. aureus, поэтому антибиотикотерапия приводит не только к снижению зуда, но и к улучшению течения заболевания. При выраженном зуде рекомендовано назначение габапентина – препарата, используемого для лечения нейропатических болей. Начинают с дозы 900 мг/день в 3 приема и постепенно увеличивают дозу до 3600 мг/день. Побочный седативный эффект позволяет пациентам нормализовать ночной сон. Для усиления снотворного эффекта в ночное время к терапии можно добавить 7, 5-15 мг миртазапина н/ночь.
5. Определение эффективности лечения
При СС используются критерии ответа на лечение, предложенные ISCL, EORTC и Американским консорциумом по кожным лимфомам (USCLC) (см. главу «Грибовидный микоз»).
CD30+ лимфопролиферативные заболевания кожи: лимфоматоидный папулез,
первичная анапластическая крупноклеточная лимфома кожи
Группа первичных кожных CD30+ лимфопролиферативных заболеваний (ЛПЗ) является второй по частоте возникновения (после грибовидного микоза) и составляет 25% всех первичных лимфом кожи. Она представляет собой спектр заболеваний, включающий лимфоматоидный папулез (ЛиП) и первичную анапластическую CD30+ крупноклеточную лимфому кожи (АКЛК).
Заболеваемость ЛиП и АКЛК в мире составляет 0, 1-0, 2 случая на 100 000 населения. ЛиП и АКЛК могут возникать во всех возрастных категориях, средний возраст дебюта заболевания для ЛиП 35-45 лет, для АКЛК 50-60 лет. Соотношение заболевших ЛиП мужчин и женщин составляет 1, 5: 1, АКЛК – 2-3: 1.
Диагностика
Диагноз CD30+ЛПЗ устанавливается на основании комплексной оценки характерной клинической картины заболевания, гистологического и иммунофенотипического исследования биоптатов из очагов поражения кожи.
Клиническое обследование пациента имеет большое значение в диагностике ЛиП, так как дает возможность не только заподозрить ЛиП, но и определить оптимальный элемент кожной сыпи для получения биоптата кожи. Для гистологического исследования рекомендовано выполнять полное удаление наиболее выраженного узелкового элемента (при ЛиП) или инцизионную биопсию узла (при АКЛК).
В большинстве случаев ЛиП характеризуется хроническим доброкачественным течением и не влияет на выживаемость, однако пациенты с ЛиП имеют высокий риск развития вторичных кожных или нодальных лимфопролиферативных заболеваний, включая грибовидный микоз (ГМ), кожную или нодальную анапластическую крупноклеточную лимфому и лимфому Ходжкина. Эти ЛиП-ассоциированные лимфомы развиваются в 4-25% случаев у пациентов с ЛиП и могут предшествовать ЛиП, возникать одновременно с ним или после его начала, что необходимо учитывать в процессе установления диагноза. АКЛК также характеризуется благоприятным прогнозом с 5-летней выживаемостью между 76% и 96%.
Таблица 7
Стадирование первичных лимфом кожи, отличных от ГМ/СС, согласно рекомендациям ISLE-EORTC
 Кожа
Кожа
Т 1 Одиночный элемент кожной сыпи
Т1а – кожный элемент < 5 см в диаметре Т1b – кожный элемент > 5 см в диаметре
Т 2 Очаговое поражение кожи: множественные высыпания, ограниченные 1 зоной или двумя рядом расположенными зонами*
Т2а – все высыпания располагаются в зоне < 15 см в диаметре
Т2b - все высыпания располагаются в зоне > 15 см < 30 см в диаметре
T2c – все высыпания располагаются в зоне > 30 см в диаметре
Т 3 Генерализованное поражение кожи
T3a – множественные высыпания, занимающие не рядом расположенные зоны
T3b – множественные высыпания, занимающие ≥ 3 зоны
Лимфатические узлы
N0 Нет увеличения периферических и центральных лимфатических узлов, их биопсия не требуется
N1 Поражение 1 группы периферических лимфатических узлов, дренирующих область настоящих или предшествующих кожных высыпаний
N2 Поражение 2 или более групп периферических лимфатических узлов, или поражение любых периферических лимфатических узлов, не дренирующих область настоящих или предшествующих кожных высыпаний
N3 Поражение центральных лимфатических узлов
Внутренние органы
M0 Нет вовлечения внутренних органов
M1 Вовлечение внутренних органов (с уточнением органа и морфологическим подтверждением)
* Деление кожного покрова на зоны приведено на схеме 2
Схема 2
Зоны кожного покрова
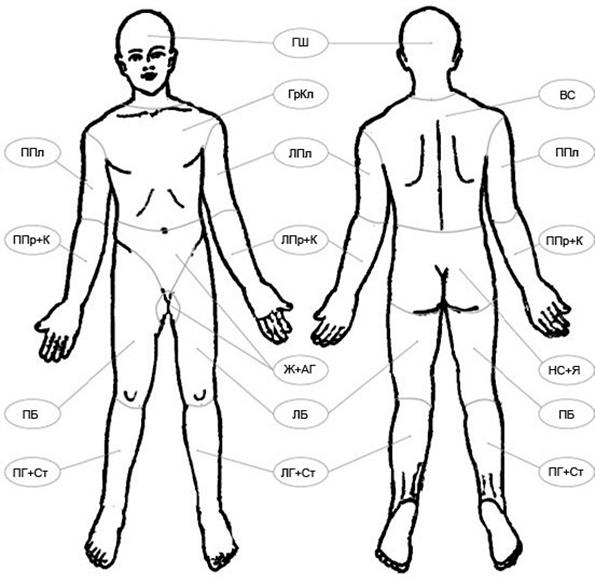 |
ГШ – голова и шея; ГрКл – грудная клетка; ВС – верхняя часть спины; НС+Я – нижняя часть спины и ягодицы; Ж+АГ – живот и аногенитальная область; ППл – правое плечо; ППр+К – правое предплечье и кисть; ЛПл – левое плечо; ЛПр+К – левое предплечье и кисть; ПБ – правое бедро; ПГ+Ст – правая голень и стопа; ЛГ+Ст – левая голень и стопа.
Для диагностики ЛиП используются следующие диагностические критерии:
1. Клинические критерии
· рецидивирующие высыпания самопроизвольно разрешающихся сгруппированных или диссеминированных папулезных элементов (под самопроизвольным разрешением понимают спонтанную регрессию каждого индивидуального элемента в течение недель или месяцев, независимо от появления новых высыпаний)
· В случае одновременного развития ЛиП и ГМ клиническая картина ЛиП будет сопровождаться появлением пятен, бляшек и узлов (в зависимости от стадии ГМ)
2. Гистологические критерии
Гистологические признаки ЛиП вариабельны и зависят от стадии развития элемента. Выделяют 4 гистологических подтипа ЛиП. Необходимо учитывать, что разные подтипы могут наблюдаться у одного и того же пациента одновременно в разных высыпаниях.
· ЛиП тип А (наиболее часто встречающийся): дермальный очаговый
«клинообразный» инфильтрат, состоящий из отдельных или сгруппированных CD30+ опухолевых клеток с примесью многочисленных малых лимфоцитов, гистиоцитов, нейтрофильных и эозинофильных лейкоцитов
· ЛиП тип В: эпидермотропный инфильтрат, состоящий из атипичных CD30+ или CD30- лимфоидных клеток малых и средних размеров с церебриформными ядрами (гистологическая картина напоминает ГМ)
· ЛиП тип С: сливающиеся поля CD30+ крупных атипичных лимфоидных клеток, примесь воспалительного инфильтрата незначительна
· ЛиП тип D: эпидермотропный инфильтрат, состоящий из атипичных CD8+ и CD30+ лимфоидных клеток малых и средних размеров (гистологическая картина напоминает первичную кожную агрессивную эпидермотропную CD8+ цитотоксическую Т-клеточную лимфому)
3. Иммуногистохимические критерии
· В большинстве случаев CD30+ опухолевые клетки экспрессируют CD4,
реже могут наблюдаться CD8+ или CD56+ фенотипы
· Т-клеточно-ассоциированные антигены (CD45RO) экспрессируются с вариабельной потерей пан-Т-клеточных антигенов (CD2, CD3, CD5)
· Необходимо учитывать, что крупные атипично выглядящие CD30+ клетки могут встречаться при различных воспалительных и инфекционных заболеваниях
Для диагностики АКЛК используются следующие диагностические критерии:
1. Клинические критерии
· солитарные, сгруппированные, или множественные высыпания
· отсутствие клинических признаков ЛиП, ГМ или других Т-клеточных лимфом кожи
· отсутствие внекожных очагов поражения
2. Гистологические критерии:
· плотный очаговый или диффузный инфильтрат, состоящий из крупных плеоморфных, анапластических клеток или клеток с иммунобластной морфологией
· в инфильтрате могут обнаруживаться скопления малых реактивных лимфоцитов и эозинофильных лейкоцитов
3. Иммуногистохимические критерии:
· экспрессия CD30 должна обнаруживаться не менее чем на 75%
опухолевых клеток
· в большинстве случаев опухолевые клетки экспрессируют CD4 или CD8 антигены с вариабельной потерей пан-Т-клеточных антигенов (CD2, CD3, CD5)
· в отличие от нодальной формы при АКЛК экспрессируется CLA (HECA- 452) и не экспрессируется EMA
· в отличие от нодальной формы экспрессия ALK-1 и t(2; 5) транслокация обычно отсутствуют при АКЛК
Стадирование и план обследования
Стадирование СD30+ЛПЗ проводится согласно рекомендациям Международного общества по лимфомам кожи и Европейской организации по изучению и лечению рака, которые были разработаны для лимфом кожи, отличных от ГМ/синдрома Сезари (ISCL-EORTC staging system for cutaneous lymphomas other than MF/SS) (табл. 1).
Проводить стадирование по схемам Ann Arbor или TNM не рекомендуется, так как пациенты с диссеминированными высыпаниями попадают в категорию IV, что подразумевает позднюю стадию, плохой прогноз и может привести к назначению неадекватного лечения.
План обследования пациентов с CD30+ ЛПЗ включает:
1. Тщательный сбор анамнеза
· рецидивирующие высыпания самопроизвольно разрешающихся сгруппированных или диссеминированных папулезных элементов (для ЛиП)
· предшествующие или сопутствующие лимфопролиферативные заболевания (болезнь Ходжкина, нодальная анапластическая крупноклеточная лимфома, ГМ)
· В-симптомы (повышение температуры тела > 380, проливные ночные поты, потеря веса более 10% за последние 6 месяцев)
2. Физикальный осмотр
· количество и размер высыпаний (наличие пятен и бляшек указывает на возможную ассоциацию с ГМ)
· идентификация пальпируемых л/у и органомегалии
3. Лабораторные исследования
· клинический и биохимический анализы крови (включая ЛДГ)
4. Радиологическое обследование
· ЛиП: рентгенография грудной клетки, УЗИ органов брюшной полости и малого таза, или компьютерная томография (для пациентов с отсутствием увеличенных л/у, гепатоспленомегалии и В-симптомов)
· АКЛК: компьютерная томография с контрастированием (грудная клетка, брюшная полость, малый таз).
5. Биопсия кожи
· гистологическое исследование, иммуногистохимическое исследование, включающее следующие маркеры: CD3, CD4, CD5, CD7, CD8, CD20, CD30, ALK-1, EMA, CLA, CD56, TIA-1, granzim B, perforin)
6. Биопсия л/у: при увеличении > 1, 5 см в диаметре и/или плотной, неравномерной консистенции
7. Трепанобиопсия костного мозга
· ЛиП: не выполняется
· АКЛК: выполняется у пациентов с множественными высыпаниями и поражением регионарных л/у
Определение эффективности лечения
ISCL, EORTC и Американским консорциумом по кожным лимфомам (USCLC) были предложены следующие критерии ответа на лечение при CD30+ЛПЗ:
1. Кожа
А. ЛиП:
Полная ремиссия ( ПР ): 100% разрешения высыпаний.
Частичная ремиссия ( ЧР ): 50%-99% разрешения высыпаний от исходного уровня, отсутствие новых более крупных узелковых высыпаний > 2см в диаметре.
Стабилизация заболевания: от < 50% увеличения до < 50% разрешения высыпаний от исходного уровня, отсутствие новых более крупных узелковых высыпаний > 2см в диаметре.
Утрата ответа: увеличение высыпаний от наименьшего уровня на 50%
исходного уровня у больных с достигнутой ПР или ЧР.
Прогрессирование заболевания: появление новых более крупных и персистирующих узелковых высыпаний > 2см в диаметре или внекожное распространение заболевания.
Рецидив: появление кожных высыпаний у пациентов в состоянии полной ремиссии заболевания.
В: АКЛК:
ПР: 100% разрешения высыпаний.
ЧР: 50%-99% разрешения высыпаний от исходного уровня, отсутствие новых узлов.
Стабилизация заболевания: от < 25% увеличения до < 50% разрешения высыпаний от исходного уровня.
Прогрессирование заболевания: увеличение высыпаний более чем на 25% от исходного уровня, или увеличение высыпаний от наименьшего уровня на 50% исходного уровня у больных с достигнутой ПР или ЧР
Рецидив: появление кожных высыпаний у пациентов в состоянии полной ремиссии заболевания.
2. Лимфатические узлы
ПР: все лимфатические узлы < 1, 5 см в наибольшем диаметре или гистологически негативные. Кроме того, лимфатические узлы, которые на момент постановки диагноза были менее 1, 5 см и при этом гистологически позитивны, должны уменьшиться до 1 см или быть гистологически негативными.
ЧР: кумулятивное снижение ≥ 50% СПР (сумма произведений максимального продольного размера × максимальный поперечный размер каждого пораженного л/у) и отсутствие новых л/у > 1, 5 см в диаметре по длинной оси или > 1 см по короткой оси
Стабилизация заболевания: отсутствие критериев для ПР и ЧР и прогрессирования заболевания
Прогрессирование заболевания: повышение ≥ 50% СПР от исходных размеров л/у, или новый л/у > 1, 5 см в диаметре по длинной оси или > 1 см по короткой оси, или отсутствие ответа: увеличение СПР на > 50% от низшего уровня у пациентов в ЧР
Рецидив: появление новых гистологически доказанных л/у > 1, 5 см в наибольшем диаметре у пациентов с ПР
3. Висцеральные органы
ПР: отсутствие увеличения органа при физикальном осмотре и отсутствие патологических изменений при томографии, биопсия любых новых очагов, появившихся после лечения, для исключения лимфомы.
ЧР: ≥ 50% регрессии очагов печени, селезенки или других изначально пораженных органах при возможности измерить объем поражения (СПР), отсутствие увеличения органа в размерах и вовлечения новых органов
Стабилизация заболевания: отсутствие критериев для ПР, ЧР и прогрессирования заболевания
Прогрессирование заболевания: > 50% увеличения органа в размере, или поражение нового органа, или отсутствие ответа: увеличение СПР на
> 50% от низшего уровня у пациентов в ЧР
Рецидив: вовлечение нового органа у пациентов с ПР
На настоящий момент остается неразрешенным вопрос, могут ли при ЛиП поражаться л/у и висцеральные органы. Возникновение CD30+ лимфопролиферативного процесса в л/у и висцеральных органах рекомендовано расценивать как ассоциированную с ЛиП вторичную анапластическую крупноклеточную лимфому.
 Лечение ЛиП 3
Лечение ЛиП 3
Лечебная тактика
Проведенные исследования эффективности разных видов лечения ЛиП показали, что на настоящий момент не существует терапии, способной изменить течение заболевания или предотвратить возникновение ЛиП- ассоциированных вторичных лимфом, поэтому тактика воздержания от активных терапевтических воздействий является предпочтительной.
 |
3 Из-за выраженной гетерогенности и низкой распространенности заболевания, количество контролируемых клинических исследований невелико, поэтому все рекомендации данного раздела имеют уровень доказательности III-IV, C-D
Учитывая прекрасный прогноз ЛиП и высокую частоту рецидивов практически после любого вида терапии большинству пациентов предлагается тактика «наблюдай и жди».
Терапия, рецидивы и последующее наблюдение
Для лечения пациентов с многочисленными и диссеминированными высыпаниями с наилучшими результатами применяется ПУВА-терапия (см. главу «Грибовидный микоз») и лечение низкими дозами метотрексата (5-30 мг/неделю, с 1-4-недельными перерывами). Оба вида терапии вызывают снижение количества и быстрое разрешение высыпаний у большинства пациентов, но полная ремиссия достигается редко, после прекращения лечения (или снижения дозы) быстро возникают рецидивы. Из-за склонности ЛиП к рецидивированию может потребоваться поддерживающая терапия для контроля течения заболевания. При этом необходимо учитывать, что длительное применение ПУВА-терапии может привести к повышенному риску возникновения рака кожи, метотрексата – к развитию фиброза печени.
У пациентов с узелковыми высыпаниями > 2см в диаметре, не разрешающихся в течение нескольких месяцев, может выполняться хирургическое удаление элементов или локальная лучевая терапия как альтернативный подход вместо тактики «наблюдай и жди».
Длительное персистирование узелковых высыпаний > 2см в диаметре без отсутствия их самостоятельного разрешения требует проведения повторной биопсии кожи для исключения вторичной анапластической крупноклеточной лимфомы.
Пациенты с ЛиП должны быть под наблюдением в течение всей жизни из-за риска развития у них вторичных лимфопролиферативных заболеваний (4-25% случаев) даже через несколько десятилетий после начала ЛиП и при отсутствии кожных высыпаний ЛиП. Рекомендованы ежегодные осмотры с проведением рентгенографии грудной клетки и УЗИ органов брюшной полости и малого таза.
 Лечение АКЛК 4
Лечение АКЛК 4
Ведение пациентов с АКЛК зависит от размера, количества и степени распространения кожных высыпаний и наличия внекожного распространения заболевания.
1. Для АКЛК с солитарными или сгруппированными высыпаниями хирургическое удаление или лучевая терапия являются предпочитаемым лечением первой линии с достижением ПР в 95% случаев. Рецидивы возникают у 40% пациентов с одинаковой частотой после обоих видов лечения. При рецидивах, ограниченных кожей, не наблюдается ухудшения прогноза, и они не требуют других видов лечения.
2. Для АКЛК с множественными распространенными высыпаниями рекомендовано лечение низкими дозами метотрексата (5-25 mg в неделю), при отсутствии эффекта можно комбинировать лечение с интерфероном- α.
3. Для АКЛК с внекожным распространением рекомендована полихимиотерапия, наиболее часто применяется режим CHOP.
Литература
1. Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sé zary syndrome: A proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007; 110: 1713-1722
2. Willemze R, Jaffe ES, Burg G, et al: WHO-EORTC classification for cutaneous lymphomas. Blood. 2005; 105: 3768-3785
3. Белоуссова И. Э., Казаков Д. В,, Криволапов Ю. А. Современные подходы к диагностике и лечению первичных лимфом кожи на основе новой ВОЗ-EORTC классификации. Т-клеточные лимфомы кожи. Архив патологии. 2007; 69(5): 11-17
4. Lutzner M, Edelson R, Schein P, et al: Cutaneous T-cell lymphomas: The Sé zary syndrome, mycosis fungoides, and related disorders. Ann Intern Med. 1975; 83: 534-552
| |
4 Из-за выраженной гетерогенности и низкой распространенности заболевания, количество контролируемых клинических исследований невелико, поэтому все рекомендации данного раздела имеют уровень доказательности III-IV, C-D
5. Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005; 53(6): 1053-63
6. Братцева Е. В., Ротанов С. В. Современные подходы к диагностике грибовидного микоза. Вестник дерматологии и венерологии. 2010; 6: 16-22
7. Kim EJ, Hess S, Richardson SK, et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J Clin Invest. 2005; 115(4): 798-812
8. Thurber SE, Zhang B, Kim YH, et al. T-cell clonality analysis in biopsy specimens from two different skin sites shows high specificity in the diagnosis of patients with suggested mycosis fungoides. J Am Acad Dermatol. 2007; 57(5): 782-90
9. Horwitz SM, Olsen EA, Duvic M, et al. Review of the treatment of mycosis fungoides and sé zary syndrome: a stage-based approach. J Natl Compr Canc Netw. 2008; 6(4): 436-42.
10. Zackheim HS. Treatment of patch-stage mycosis fungoides with topical corticosteroids. Dermatol Ther. 2003; 16(4): 283-7
11. Diederen PV, van Weelden H, Sanders CJ, et al. Narrowband UVB and psoralen-UVA in the treatment of early-stage mycosis fungoides: a retrospective study. J Am Acad Dermatol. 2003; 48(2): 215-9
12. Gathers RC, Scherschun L, Malick F, et al. Narrowband UVB phototherapy for early-stage mycosis fungoides. J Am Acad Dermatol. 2002; 47(2): 191-7
13. Ponte P, Serrã o V, Apetato M. Efficacy of narrowband UVB vs. PUVA in patients with early-stage mycosis fungoides. J Eur Acad Dermatol Venereol. 2010; 24(6): 716-21
14. Querfeld C, Rosen ST, Kuzel TM, et al. Long-term follow-up of patients with early-stage cutaneous T-cell lymphoma who achieved complete remission with psoralen plus UV-A monotherapy. Arch Dermatol. 2005; 141(3): 305-11
15. Hoppe RT. Mycosis fungoides: radiation therapy. Dermatol Ther. 2003; 16(4): 347-54
16. Hymes KB. Choices in the treatment of cutaneous T-cell lymphoma. Oncology (Williston Park). 2007; 21(2 Suppl 1): 18-23
17. Keehn CA, Belongie IP, Shistik G, et al. The diagnosis, staging, and treatment options for mycosis fungoides. Cancer Control. 2007; 14(2): 102-11
18. Zhang C, Duvic M. Treatment of cutaneous T-cell lymphoma with retinoids. Dermatol Ther. 2006; 19(5): 264-71
19. Olsen EA. Interferon in the treatment of cutaneous T-cell lymphoma. Dermatol Ther. 2003; 16(4): 311-21
20. Zackheim HS, Kashani-Sabet M, McMillan A. Low-dose methotrexate to treat mycosis fungoides: a retrospective study in 69 patients. J Am Acad Dermatol. 2003; 49(5): 873-8
21. Duvic M, Talpur R, Ni X, Zhang C, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2007; 109(1): 31-9
22. Olsen EA, Kim YH, Kuzel TM, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007; 25(21): 3109-15
23. Duvic M, Olsen EA, Breneman D, et al. Evaluation of the long-term tolerability and clinical benefit of vorinostat in patients with advanced cutaneous T-cell lymphoma. Clin Lymphoma Myeloma. 2009; 9(6): 412-6
24. Wu PA, Kim YH, Lavori PW, et al. A meta-analysis of patients receiving allogeneic or autologous hematopoietic stem cell transplant in mycosis fungoides and Sé zary syndrome. Biol Blood Marrow Transplant. 2009; 15(8): 982-90
25. Duarte RF, Canals C, Onida F, et al. Allogeneic hematopoietic cell transplantation for patients with mycosis fungoides and Sé zary syndrome: a retrospective analysis of the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol. 2010; 28(29): 4492-9
26. Prince HM, Whittaker S, Hoppe RT. How I treat mycosis fungoides and Sé zary syndrome. Blood. 2009; 114: 4337-53
27. Molin L, Thomsen K, Volden G, et al. Combination chemotherapy in the tumor stage of mycosis fungoides with cyclophosphamide, vincristine, vp-16, adriamycin and prednisolone (COP, CHOP, CAVOP): a report from the Scandinavian mycosis fungoides study group. Acta Derm Venereol. 1980; 60(6): 542-544
28. Akpek G, Koh HK, Bogen S, O’Hara C, Foss FM. Chemotherapy with etoposide, vincristine, doxorubicin, bolus cyclophosphamide, and oral prednisone in patients with refractory cutaneous T-cell lymphoma. Cancer. 1999; 86(7): 1368-1376
29. Duvic M, Apisarnthanarax N, Cohen DS, et al. Analysis of long-term outcomes of combined modality therapy for cutaneous T-cell lymphoma. J Am Acad Dermatol. 2003; 49(1): 35-49
30. Wollina U, Dummer R, Brockmeyer NH, et al. Multicenter study of pegylated liposomal doxorubicin in patients with cutaneous T-cell lymphoma. Cancer. 2003; 98(5): 993-1001
31. Tsimberidou AM, Giles F, Duvic M, et al. Phase II study of pentostatin in advanced T-cell lymphoid malignancies: update of an M. D. Anderson Cancer Center series. Cancer. 2004; 100(2): 342-349
32. Foss FM, Ihde DC, Linnoila IR, et al. Phase II trial of fludarabine phosphate and interferon alfa-2a in advanced mycosis fungoides/Sezary syndrome. J Clin Oncol. 1994; 12(10): 2051- 2059
33. Scarisbrick JJ, Child FJ, Clift A, et al. A trial of fludarabine and cyclophosphamide combination chemotherapy in the treatment of advanced refractory primary cutaneous T- cell lymphoma. Br J Dermatol. 2001; 144(5): 1010-1015
34. Zinzani PL, Baliva G, Magagnoli M, et al. Gemcitabine treatment in pretreated cutaneous T-cell lymphoma: experience in 44 patients. J Clin Oncol. 2000; 18(13): 2603-2606
35. Olsen EA, Whittaker S, Kim YH, et al. Clinical end points and response criteria in mycosis fungoides and Sé zary syndrome: a consensus statement of the International Society for Cutaneous Lymphomas, the United States Cutaneous Lymphoma Consortium, and the Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. J Clin Oncol. 2011; 29: 2598-607
36. Kempf W, Willemze R, Jaffe ES, et al. CD30+ T-cell lymphoproliferative disorders. In: LeBoit P, Burg G, Weedon D, Sarasin A, eds. World Health Organization Classification of Tumours: Pathology and Genetics of Skin Tumours. Lyon, France: IARC Press; 2006; 179- 181.
37. Ralfkiaer E, Willemze R, Paulli M, Kadin ME. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. In: Swerdlow SH, Campo E, Harris NL, et al., eds. World
Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues (4th ed). Lyon, France: IARC Press; 2008; 300-301.
38. Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma
39. Guitart J, Querfeld C. Cutaneous CD30 lymphoproliferative disorders and similar conditions: a clinical and pathologic prospective on a complex issue. Semin Diagn Pathol. 2009; 26(3): 131-140.
40. Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007; 110(2): 479-484.
41. Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the longterm follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000; 95(12): 3653-3661.
42. Liu HL, Hoppe RT, Kohler S, et al. CD30+cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003; 49(6): 1049-1058.
43. Wantzin GL, Thomsen K. PUVA-treatment in lymphomatoid papulosis. Br J Dermatol. 1982; 107(6): 687-690.
44. Lange Wantzin G, Thomsen K. Methotrexate in lymphomatoid papulosis. Br J Dermatol. 1984; 111(1): 93-95.
45. Fujita H, Nagatani T, Miyazawa M, et al. Primary cutaneous anaplastic large cell lymphoma successfully treated with low-dose oral methotrexate. Eur J Dermatol. 2008; 18(3): 360-361.
|
|
|
© helpiks.su При использовании или копировании материалов прямая ссылка на сайт обязательна.
|