- Автоматизация
- Антропология
- Археология
- Архитектура
- Биология
- Ботаника
- Бухгалтерия
- Военная наука
- Генетика
- География
- Геология
- Демография
- Деревообработка
- Журналистика
- Зоология
- Изобретательство
- Информатика
- Искусство
- История
- Кинематография
- Компьютеризация
- Косметика
- Кулинария
- Культура
- Лексикология
- Лингвистика
- Литература
- Логика
- Маркетинг
- Математика
- Материаловедение
- Медицина
- Менеджмент
- Металлургия
- Метрология
- Механика
- Музыка
- Науковедение
- Образование
- Охрана Труда
- Педагогика
- Полиграфия
- Политология
- Право
- Предпринимательство
- Приборостроение
- Программирование
- Производство
- Промышленность
- Психология
- Радиосвязь
- Религия
- Риторика
- Социология
- Спорт
- Стандартизация
- Статистика
- Строительство
- Технологии
- Торговля
- Транспорт
- Фармакология
- Физика
- Физиология
- Философия
- Финансы
- Химия
- Хозяйство
- Черчение
- Экология
- Экономика
- Электроника
- Электротехника
- Энергетика
PREPARATORY PROBLEMS: THEORETICAL
19th – 29th July, 2018
Bratislava, SLOVAKIA
Prague, CZECH REPUBLIC
www. 50icho. eu
PREPARATORY PROBLEMS: THEORETICAL
SOLUTIONS
| | 50th IChO 2018 International Chemistry Olympiad SLOVAKIA & CZECH REPUBLIC BACK TO WHERE IT ALL BEGAN |
NOT TO BE MADE ACCESSIBLE BEFORE 1st JUNE, 2018
Table of Contents
Problem 1. Synthesis of hydrogen cyanide. 2
Problem 2. Thermochemistry of rocket fuels. 3
Problem 3. HIV protease. 6
Problem 4. Enantioselective hydrogenation. 8
Problem 5. Ultrafast reactions. 9
Problem 6. Kinetic isotope effects. 13
Problem 7. Designing a photoelectrochemical cell 14
Problem 8. Fuel cells. 16
Problem 9. Acid-base equilibria in blood. 18
Problem 10. Ion exchange capacity of a cation exchange resin. 20
Problem 11. Weak and strong cation exchange resin. 21
Problem 12. Uranyl extraction. 22
Problem 13. Determination of active chlorine in commercial products. 24
Problem 14. Chemical elements in fireworks. 25
Problem 15. Colours of complexes. 27
Problem 16. Iron chemistry. 29
Problem 17. Cyanido- and fluorido-complexes of manganese. 34
Problem 18. The fox and the stork. 37
Problem 19. Structures in the solid state. 39
Problem 20. Cyclobutanes. 41
Problem 21. Fluorinated radiotracers. 42
Problem 22. Where is lithium?. 44
Problem 23. Synthesis of eremophilone. 45
Problem 24. Cinnamon all around. 46
Problem 25. All roads lead to caprolactam.. 48
Problem 26. Ring opening polymerizations (ROP) 50
Problem 27. Zoniporide. 52
Problem 28. Nucleic acids. 54
Problem 1. Synthesis of hydrogen cyanide
1. 1 Degussa process (BMA process):
Δ rHm = − Δ fHm(CH4) − Δ fHm(NH3) + Δ fHm(HCN) + 3 Δ fHm(H2)
Δ rHm = [− (− 90. 3) − (− 56. 3) + 129. 0 + 3 × 0] kJ mol− 1 = 275. 6 kJ mol− 1
Andrussow process:
Δ rHm = − Δ fHm(CH4) − Δ fHm(NH3) − 3/2 Δ fHm(O2) + Δ fHm(HCN) + 3 Δ fHm(H2O)
Δ rHm = [− (− 90. 3) − (− 56. 3) − 3/2 × 0 + 129. 0 + 3 × (− 250. 1)] kJ mol− 1 = − 474. 7 kJ mol− 1
1. 2 An external heater has to be used in the Degussa process (BMA process) because
the reaction is endothermic.
1. 3 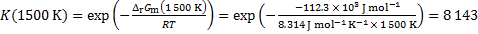

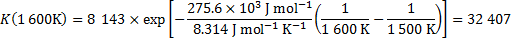
The result is in accordance with the Le Chatelier’s principle because the reaction is endothermic and therefore an increase in temperature shifts the equilibrium toward products (in other words, the equilibrium constant increases).
1. 4 The equilibrium constant of the reaction in the Andrussow process decreases with
an increase in temperature because the reaction is exothermic.
Problem 2. Thermochemistry of rocket fuels
Notation of indexes: 0M – hydrazine, 1M – methylhydrazine, 2M – 1, 1-dimethylhydrazine
Standard conditions: T° = 298. 15 K; p° = 101 325 Pa.
All values given below are evaluated from non-rounded intermediate results.
2. 1 Calculation of the number of moles corresponding to 1 g of the samples: ni = mi / Mi
M0M = 32. 05 g mol− 1; M1M = 46. 07 g mol− 1; M2M = 60. 10 g mol− 1.
n0M = 31. 20 mmol; n1M = 21. 71 mmol; n2M = 16. 64 mmol.
Calculation of combustion heat: qi = Ccal × Δ Ti
q0M = 16. 83 kJ; q1M = 25. 60 kJ; q2M = 30. 11 kJ.
Calculation of the molar internal energies of combustion: Δ cUi = − qi / ni
Δ combU0M = − 539. 40 kJ mol− 1; Δ combU1M = − 1 179. 48 kJ mol− 1;
Δ combU2M = − 1 809. 64 kJ mol− 1.
Bomb calorimeter combustion reactions with the stoichiometric coefficients added:
Hydrazine N2H4 (l) + O2 (g) → N2 (g) + 2 H2O (g)
Methylhydrazine N2H3CH3 (l) + 2. 5 O2 (g) → N2 (g) + CO2 (g) + 3 H2O (g)
1, 1-Dimethylhydrazine N2H2(CH3)2 (l) + 4 O2 (g) → N2 (g) + 2 CO2 (g) + 4 H2O (g)
Calculation of the molar enthalpies of combustion: Δ cHi = Δ cUi + Δ cn(gas)RTstd
Δ combH0M = − 534. 44 kJ mol− 1; Δ combH1M = − 1 173. 29 kJ mol− 1;
Δ combH2M = − 1 802. 20 kJ mol− 1.
2. 2 Calculation of the molar enthalpies of formation:
Δ formH0M = 2 Δ formHH2O, g − Δ combH0M = +50. 78 kJ mol− 1
Δ formH1M = 3 Δ formHH2O, g + Δ formHCO2 − Δ combH1M = +54. 28 kJ mol− 1
Δ formH2M = 4 Δ formHH2O, g + 2 Δ formHCO2 − Δ combH2M = +47. 84 kJ mol− 1
Rocket engines combustion reactions:
Hydrazine N2H4 (l) + 1/2 N2O4 (l) → 2 H2O (g) + 3/2 N2 (g)
Methylhydrazine N2H3CH3 (l) + 5/4 N2O4 (l) → CO2 (g) + 3 H2O (g) + 9/4 N2 (g)
1, 1-Dimethylhydrazine N2H2(CH3)2 (l) + 2 N2O4 (l) → 3 N2 (g) + 4 H2O (g) + 2 CO2 (g)
Calculation of molar reaction enthalpies, related to one mole of hydrazine derivatives:
Δ reH0M = (2 Δ formHH2O, g − 1/2 Δ formHN2O4 − Δ formH0M) = − 538. 98 kJ mol− 1
Δ reH1M = (Δ formHCO2 + 3 Δ formHH2O, g − 5/4 Δ formHN2O4 − Δ formH1M) = − 1 184. 64 kJ mol− 1
Δ reH2M = (2 Δ formHCO2 + 4 Δ formHH2O, g − 2 Δ formHN2O4 − 1 Δ formH2M) = − 1 820. 36 kJ mol− 1
2. 3 Calculation of the standard molar reaction enthalpies, related to one mole of hydrazine derivatives:
Δ reH°0M = Δ reH0M − 2 Δ vapHH2O = − 620. 28 kJ mol− 1
Δ reH°1M = Δ reH1M − 3 Δ vapHH2O = − 1 306. 59 kJ mol− 1
Δ reH°2M = Δ reH2M − 4 Δ vapHH2O = − 1 982. 96 kJ mol− 1
Calculation of the standard molar reaction entropies, related to one mole of hydrazine derivatives:
Δ reS°0M = (2 SH2O, l + 3/2 SN2 − 1/2 SN2O4 − S0M) = 200. 67 J K− 1 mol− 1
Δ reS°1M = (SCO2 + 3 SH2O, l + 9/4 SN2 − 5/4 SN2O4 − S1M) = 426. 59 J K− 1 mol− 1
Δ reS°2M = (2 SCO2 + 4 SH2O, l + 3 SN2 − 2 SN2O4 − S2M) = 663. 69 J K− 1 mol− 1
Calculation of standard molar reaction Gibbs energies:
Δ reG°0M = Δ reH°0M − T° × Δ reS°0M = − 680. 11 kJ mol− 1
Δ reG°1M = Δ reH°1M − T° × Δ reS°1M = − 1 433. 77 kJ mol− 1
Δ reG°2M = Δ reH°2M − T° × Δ reS°2M = − 2 180. 84 kJ mol− 1
Estimation of the equilibrium constants for combustion reactions:
Ki = exp(− Δ reG°i / (RT°))
K0M = e274. 37 ≈ 1 × 10119
K1M = e578. 41 ≈ 1 × 10251
K2M = e879. 79 ≈ 1 × 10382
Equilibrium constants are practically equal to infinity; the equilibrium mixture of the outlet gases contains reaction products only.
2. 4 All reactions increase the number of the moles of gaseous species, so increasing the pressure will suppress the extent of the reaction (though negligibly for such values of K). All reactions are strongly exothermic, so increasing the temperature will affect the equilibrium in the same direction as pressure.
2. 5 Summarizing the chemical equation representing the fuel mixture combustion:
N2H4 (l) + N2H3CH3 (l) + N2H2(CH3)2 (l) + 3. 75 N2O4 (l) → 6. 75 N2 (g) + 9 H2O (g) + 3 CO2 (g)
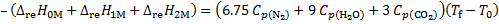 , solve for Tf
, solve for Tf
Tf = 4 288. 65 K
2. 6 Burning of 1, 1-dimethylhydrazine with oxygen can be expressed as:
N2H2(CH3)2 (l) + 4 O2 (g) → N2 (g) + 2 CO2 (g) + 4 H2O (g)
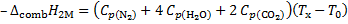 , solve for Tx
, solve for Tx
Tx = 5 248. 16 K
2. 7 There is no temperature range of coexistence of both liquid oxygen and
1, 1-dimethylhydrazine, either 1, 1-dimethylhydrazine is liquid and O2 is a supercritical fluid, or O2 is liquid and 1, 1-dimethylhydrazine is solid.
2. 8 Very high working temperatures maximize the temperature difference term in relation to the hypothetical efficiency of the Carnot engine. Assuming the low temperature equals T°, we get: η = (Tf − T°) / Tf = 93. 0%.
Problem 3. HIV protease
3. 1 Lopinavir binds most strongly, as illustrated by its smallest dissociation constant KD.
3. 2 Apply Δ G° = − RT lnKD, and consider that the dissociation and the binding are opposite reactions. Thus, Δ G°(bind. ) = − Δ G°(dissoc. ) = RT lnKD, or in a slightly different way, Δ G°(bind. ) = − RT lnKA = − RT ln[1 / KD] = RT lnKD. See below for the numerical results.
3. 3 Consider Δ G° = Δ H° − TΔ S°. Thus, perform a linear regression of the temperature dependence of Δ G°. This can be done in at least two simplified ways: (i) Plot the dependence and draw a straight line connecting the four data points in the best way visually. Then, read off the slope and intercept of the straight line, which correspond to − Δ S° and Δ H°, respectively. (ii) Alternatively, choose two data points and set up and solve a set of two equations for two unknowns, which are Δ S° and Δ H°. The most accurate result should be obtained if the points for the lowest and highest temperatures are used. See below for the numerical results.
| Temperature | Amprenavir | Indinavir | Lopinavir | ||||
| °C | K | KD | Δ G° | KD | Δ G° | KD | Δ G° |
| nM | kJ mol− 1 | nM | kJ mol− 1 | nM | kJ mol− 1 | ||
| 278. 15 | 1. 39 | − 47. 2 | 3. 99 | − 44. 7 | 0. 145 | − 52. 4 | |
| 288. 15 | 1. 18 | − 49. 3 | 2. 28 | − 47. 7 | 0. 113 | − 54. 9 | |
| 298. 15 | 0. 725 | − 52. 2 | 1. 68 | − 50. 1 | 0. 101 | − 57. 1 | |
| 308. 15 | 0. 759 | − 53. 8 | 1. 60 | − 51. 9 | 0. 0842 | − 59. 4 | |
| Δ S° | kJ K− 1 mol− 1 | 0. 228 | 0. 239 | 0. 233 | |||
| Δ H° | kJ mol− 1 | 16. 3 | 21. 5 | 12. 4 | |||
| coeff. of determin. | 0. 990 | 0. 989 | 0. 999 | ||||
| Temperature | Nelfinavir | Ritonavir | Saquinavir | ||||
| °C | K | KD | Δ G° | KD | Δ G° | KD | Δ G° |
| nM | kJ mol− 1 | nM | kJ mol− 1 | nM | kJ mol− 1 | ||
| 278. 15 | 6. 83 | − 43. 5 | 2. 57 | − 45. 7 | 0. 391 | − 50. 1 | |
| 288. 15 | 5. 99 | − 45. 4 | 1. 24 | − 49. 1 | 0. 320 | − 52. 4 | |
| 298. 15 | 3. 67 | − 48. 1 | 0. 831 | − 51. 8 | 0. 297 | − 54. 4 | |
| 308. 15 | 2. 83 | − 50. 4 | 0. 720 | − 53. 9 | 0. 245 | − 56. 7 | |
| Δ S° | kJ K− 1 mol− 1 | 0. 236 | 0. 273 | 0. 218 | |||
| Δ H° | kJ mol− 1 | 22. 4 | 29. 8 | 10. 5 | |||
| coeff. of determin. | 0. 995 | 0. 989 | 0. 999 | ||||
Note 1: Δ S° and Δ H° may also be obtained from a fit of KD or KA, without considering Δ G°. Here, a straight line would be fitted to the dependence:
ln KA = − lnKD = Δ S° / R − Δ H° / R × 1/T.
Note 2: It is evident that the binding is entropy-driven for all the inhibitors. The entropic gain stems from the changes in the flexibility of both the protease and the inhibitors, and also involves solvent effects. However, a molecular picture of those changes is rather complex.
3. 4 The slowest dissociation is observed for the compound with the smallest dissociation rate constant, i. e. Saquinavir.
3. 5 Using the relation for the dissociation constant KD = kD / kA and the data at 25 °C, we obtain for Amprenavir: kA = kD / KD = 4. 76 × 10− 3 s− 1 / (0. 725 × 10− 9 mol L− 1) = 6. 57 × 106 L mol− 1 s− 1. Analogous calculations performed for the other inhibitors yield the following numerical results. The fastest association is exhibited by the compound with the largest association rate constant, i. e. Amprenavir.
| Amprenavir | Indinavir | Lopinavir | Nelfinavir | Ritonavir | Saquinavir | ||
| kA | 6. 57 × 106 | 2. 05 × 106 | 6. 48 × 106 | 0. 59 × 106 | 3. 12 × 106 | 1. 43 × 106 | |
| dm3 mol–1 s–1 | |||||||
3. 6 The Arrhenius equation for the rate constant reads k = A × exp[− Δ G‡ / RT]. For two known rate constants of dissociation k1 and k2 determined at temperatures T1 and T2, respectively, we obtain a system of two equations,
k1 = A × exp[− Δ G‡ / RT1]
k2 = A × exp[− Δ G‡ / RT2],
from which the activation energy of dissociation results as Δ G‡ = (ln k1 / k2) / (1 / RT2 − 1 / RT1). Numerically, the activation energy is 8. 9 kJ mol− 1 for Lopinavir, 32. 6 kJ mol− 1 for Amprenavir (which has the fastest association rate constant) and 36. 8 kJ mol− 1 for Saquinavir (which has the lowest dissociation rate constant).
3. 7 No, these are two different compounds. The strongest protease binder is not the same inhibitor as the one with the slowest dissociation. This observation may seem
counter-intuitive if the distinction between thermodynamics (here, the strength of binding expressed by the equilibrium constant) and kinetics (the rate of binding represented by the rate constant or activation energy for dissociation) is not understood properly. While the equilibrium constant of dissociation captures the thermodynamic stability of the respective protein–inhibitor complex, the rate constant describes the kinetics of the process. These are two different sets of properties and they only become related if the rates of both dissociation and association are considered, KD = kD / kA.
Problem 4. Enantioselective hydrogenation
4. 1 Structure:
4. 2 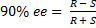 =>
=> 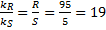 =>
=> 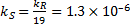 s–1
s–1
4. 3 From the previous question; at − 40 °C  . Substitute from the Arrhenius equation:
. Substitute from the Arrhenius equation:
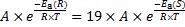 Therefore:
Therefore:

4. 4 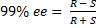 =>
=> 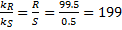
At any given temperature T:
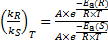 (‡) Therefore:
(‡) Therefore:
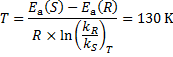
At this temperature, the reaction is likely to be really slow which would prevent its actual use.
4. 5 The main difference is that (S)-CAT will provide the (S)-product. We will do all the calculations for (R)-CAT and just invert the sign at the end. It should be noted that the amount of catalyst does not influence the enantiomeric excess; it only accelerates the reaction.
From equation (‡):
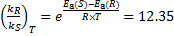 => ee = 85%
=> ee = 85%
For 90% ee: [α ]D20 (c 1. 00, EtOH) = +45°,
which means [α ]D20 (c 1. 00, EtOH) = +42. 5° for 85% ee
The same conditions are used for the measurement of the specific rotation, namely, the temperature, solvent, concentration and wavelength of the light used. Therefore, we can just invert the sign to obtain the result for the (S)-product:
[α ]D20 (c 1. 00, EtOH) = − 42. 5° = − 43°
Note: The specific rotation should be formally stated in ° dm− 1 cm3 g− 1, but in most of the current scientific literature this is simplified to ° only.
4. 6 Since the product is crystalline, the easiest method would be recrystallization. Different chiral resolution methods can also be used, for examplecrystallization with a chiral agent or separation by HPLC with a chiral stationary phase.
Problem 5. Ultrafast reactions
Note: In all equilibrium constants considered below, the concentrations should be in principle replaced by activities 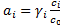 , where we use the standard state for the solution
, where we use the standard state for the solution 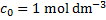 . In all calculations we assume that
. In all calculations we assume that  and for clarity, we also ignore the unity
and for clarity, we also ignore the unity 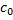 factor. We also skip the units of quantities in the intermediate steps of the calculations to make the solution easier to follow.
factor. We also skip the units of quantities in the intermediate steps of the calculations to make the solution easier to follow.
5. 1 The equilibrium constant of neutralization is given as
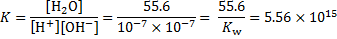
The constant K is related to the free energy change of the reaction:
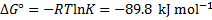
Note that the Gibbs free energy change calculated in this way corresponds to the standard state for all species, including the water solvent. The Gibbs free energy change can be expressed via the enthalpy and entropy change for the reaction

from which
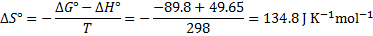
5. 2 To estimate the pH of boiling water we need to evaluate Kw at 373 K using the van ’t Hoff’s formula (alternatively, we could recalculate the constant K). Note that 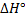 was defined for a reverse reaction, here we have to use
was defined for a reverse reaction, here we have to use 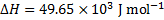 . The temperature change is given as
. The temperature change is given as

After substitution
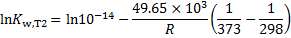
we get

which translates into proton concentration at the boiling point of water
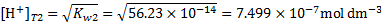
or pH
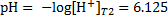
5. 3 pD is analogical to pH, i. e. pD = − log 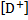 . The concentration of cations at 298 K is given as
. The concentration of cations at 298 K is given as
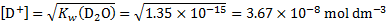
and pD is given by

5. 4

5. 5 We start from the rate equation derived in 5. 4
All concentrations can be expressed via the quantity x

Expanding the right hand side of the equation, we get

Using the equality of the backward and forward reaction rates at equilibrium

and neglecting the (small) quadratic term x2, we can rewrite the equation as

5. 6 The relaxation time is given as

At equilibrium, the backward and forward reaction rates are the same. The concentration of heavy water  is given as
is given as

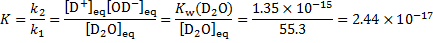
The relaxation time is then given as
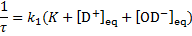
Substituting the values of all quantities
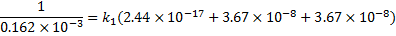
we get
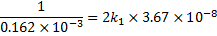
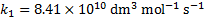
We get k2 from the equilibrium constant K
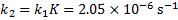
5. 7 The pH before irradiation is calculated from the dissociation constant of the ground state of 6-hydroxynaphthalene-2-sulfonate.
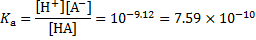
where 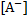 is the concentration of 6-oxidonaphthalene-2-sulfonate and
is the concentration of 6-oxidonaphthalene-2-sulfonate and 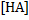 is the concentration of 6-hydroxynaphthalene-2-sulfonate.
is the concentration of 6-hydroxynaphthalene-2-sulfonate.
The concentration of 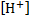 is equal to the concentration of due to electroneutrality and can be denoted as y. The equilibrium concentration of the undissociated acid is
is equal to the concentration of due to electroneutrality and can be denoted as y. The equilibrium concentration of the undissociated acid is 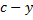 , where
, where  is the analytical concentration of the acid. The equilibrium constant is then given as
is the analytical concentration of the acid. The equilibrium constant is then given as
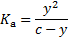
Because the amount of dissociated acid is very small, we can neglect y in the denominator
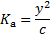
From which
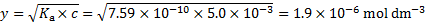

During irradiation, 1 cm3 of sample absorbs 2. 228 × 10− 3 J of energy. 1 dm3 would thus absorb 2. 228 J. The number of absorbed photons corresponds to the number of excited molecules of 6-hydroxynaphthalene-2-sulfonate.
One photon has energy
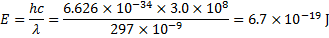
The number of absorbed photons in 1 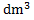 is
is
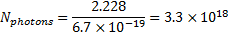
The number of moles of excited molecules of 6-hydroxynaphthalene-2-sulfonate is

The pH can again be calculated from the p 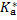 in the excited state; the analytical concentration c* of the excited acid is now
in the excited state; the analytical concentration c* of the excited acid is now 
Let us denote by x the proton concentration 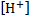 and by y* the concentration of the
and by y* the concentration of the
6-oxidonaphthalene-2-sulfonate in the excited state 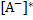 . The electroneutrality condition implies
. The electroneutrality condition implies
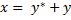
The two equilibrium constants are expressed as
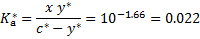
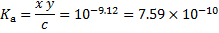
where we assumed  in the denominator of the last equation. These three equations constitute a system of equations from which we get
in the denominator of the last equation. These three equations constitute a system of equations from which we get

or
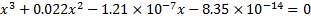
We can solve this equation e. g. with any on-line solver of cubic equations
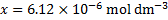
Which corresponds to

It is possible to avoid solving cubic equations by an iterative solution. In the first step, we assume that 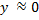 . The equation for then transforms to
. The equation for then transforms to
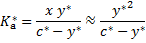
y* can be calculated from the quadratic equation
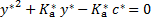
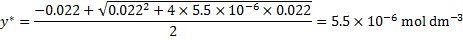
Next, we update the concentration of the anion in the ground state y from the corresponding equilibrium constant
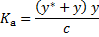
From which y can be obtained by solving a quadratic equation
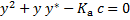
This again leads to the quadratic equation
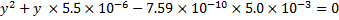

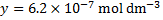
The concentration of is
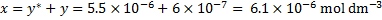
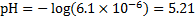
We could now repeat the whole cycle: with the first estimate of x, we would get a new value of  and continue with these new values of y and x untilconvergence is reached. At the level of precision in our calculations, the concentration is already converged in the first iteration. Generally, more iterative cycles would be needed.
and continue with these new values of y and x untilconvergence is reached. At the level of precision in our calculations, the concentration is already converged in the first iteration. Generally, more iterative cycles would be needed.
Problem 6. Kinetic isotope effects
6. 1 Reduced mass: 
Wavenumber: 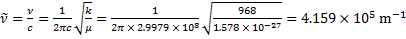
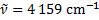
Energies: 

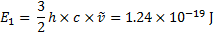
6. 2 We are going to determine the atomic mass A of the lighter isotope of the element X.
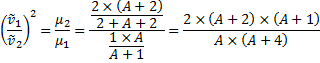
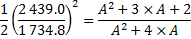
A = 79. 4 amu
A = 79; A + 2 = 81; X = Br
The second root of the quadratic equation 2. 155, which would correspond to A = 2
and A + 2 = 4, is unphysical.
6. 3 The difference of the activation energies Ea(H− C) − Ea(D− C) is equal to the negatively taken difference of zero-point vibrational energies: − E0(H− C) + E0(D− C)
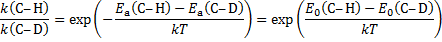

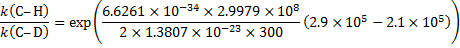
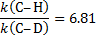
6. 4 E2 elimination. The value of the kinetic isotope effect of 6. 5 indicates that the C− H/D bond is broken in the rate-determining step of the reaction.
6. 5 For a tertiary substrate, we can expect E1 elimination, where the C− H/D bond is not broken during the rate-determining step. Therefore, we observe only a small secondary kinetic isotope effect with the kH / kD ratio slightly larger than 1. 0.
Problem 7. Designing a photoelectrochemical cell
7. 1 Reduction potentials for reactions b), c), d), f) and h) are dependent on pH.
7. 2 The potential dependence on pH is a linear function with intercept equal to E° and slope equal to:
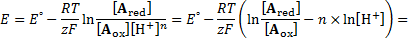
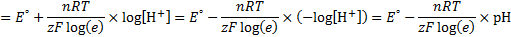
7. 3 Standard potential E°( C ) is more positive than E°( B ), hence substance C is a stronger oxidizer and will therefore oxidize substance B (a), and the standard reaction potential 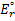 will be 0. 288V (b):
will be 0. 288V (b):

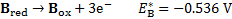
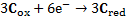
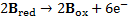
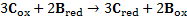
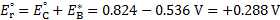
c) using formula:
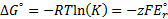
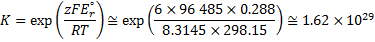
7. 4 Reaction E is pH-dependent and its potential drop is 52 mV per pH unit (as can be calculated from formula derived in question 7. 2: z = 1, n = 1, T = 262 K). The reaction potential Er= EE – ED is calculated from the equilibrium constant:
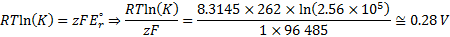
The potential for the reduction of substance E is EE = ED + Er = 0. 55 V + 0. 28 V = 0. 83 V. This value of potential is achieved at pH = 2. 31. The two lines cross at pH = 7. 7 (roughly); D will oxidize E in the pH range from 7. 7 to 13.
7. 5 Using the formula for electrolysis:

7. 6 Only materials G and I can be used to catalyze the given reaction, because their HOMOs lie below Eox and their LUMOs are higher than Ered. While material G can be irradiated only by UV light with a wavelength lower than 388 nm, material I can be irradiated by either visible or UV light, because the maximal wavelength that can be used to overcome the energy difference of 2 eV is equal to 620 nm.
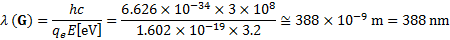
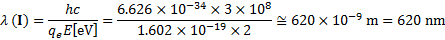
Problem 8. Fuel cells
8. 1 First, find the driving force, i. e., the Gibbs energy of the reaction H2 + ½ O2 → H2O
under standard conditions (298 K and 1 bar). Then, convert it to the EMF (voltage).
The standard reaction enthalpy and entropy are

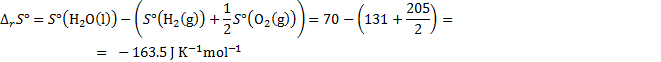
The standard change of Gibbs energy is
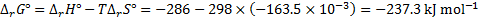
The standard EMF is then
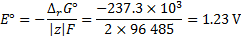
8. 2 The solution is similar to the previous one with the difference of water state.
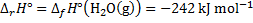
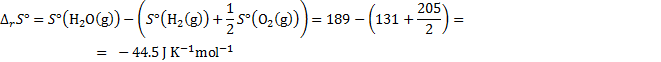
The standard change of Gibbs energy is

The standard EMF is then
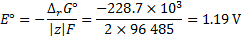
8. 3 The ideal thermodynamic efficiency is:
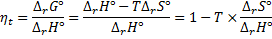
For both cells and for various temperatures, we get:
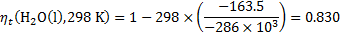
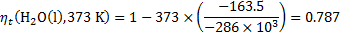
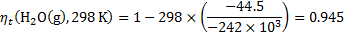
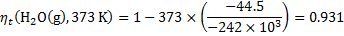
8. 4 Cathode: O2 + 4 e− + 4 H+ → 2 H2O
Anode: C4H10 + 8 H2O → 4 CO2 + 26 H+ + 26 e−
8. 5 The overall reaction is:
2 C4H10 + 13 O2 → 8 CO2 + 10 H2O
The reaction as accompanied by the transfer of 52 electrons. Hence, at standard temperature:
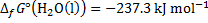
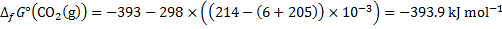
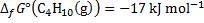
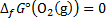

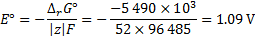
8. 6 The ideal thermodynamic efficiency is determined as:
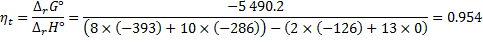
8. 7 It is the same as in the previous answer. The overall reaction is the same.
8. 8 Anode: CH3OH + H2O → 6 H+ + 6 e− + CO2
Cathode: O2 + 4 H+ + 4 e− → 2 H2O
Overall: 2 CH3OH + 3 O2 → 2 CO2 + 4 H2O
8. 9 Nernst equation
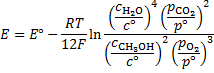
Any answer with correctly expressed activities (e. g. using molar fractions) is assumed to be correct.
8. 10 We use van ’t Hoff equation, in which we substitute EMFs for equilibrium constants. We obtain reaction enthalpy and Gibbs free energy changes, which we use to calculate the entropy change:
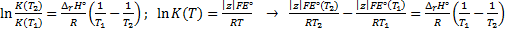

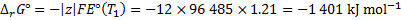
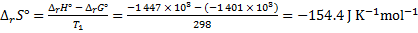
Problem 9. Acid-base equilibria in blood
9. 1 CO2 concentration:
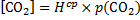


The initial concentration of bicarbonate in blood with no acid added, c(HCO3− , 37 °C):
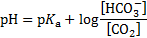
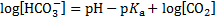
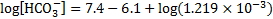
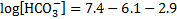
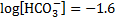
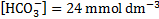
pH after 10 mmol of acids were added to 1 dm3 of the buffer solution:
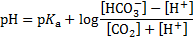
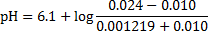

9. 2
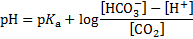


The buffering capacity of the bicarbonate buffer is higher when the system is open. However, pH is still outside the physiologic range (pH = 7. 36–7. 44). Non-bicarbonate buffers (e. g. albumin, phosphate, haemoglobin) that are present in blood additionally increase the overall buffering capacity of blood and help to keep pH within the physiologic range.
9. 3 The van ’t Hoff’s equation will be used:
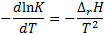
First, the integrated form is applied to calculate the reaction enthalpy from the pKa values at 37 °C and 25 °C.

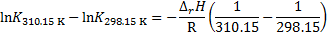
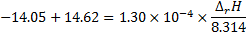
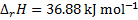
Then, that same equation is used to calculate the pKa at 20 °C:

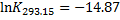


Henry’s solubility of CO2 is recalculated in an analogous way:

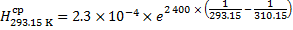
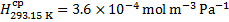
Finally, the pH of blood at 20 °C is obtained using these recalculated values:
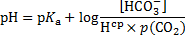
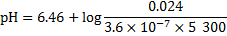
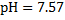
9. 4 In a working muscle, high oxygen supply is ensured by lowering the affinity towards oxygen in an acidic environment. In lungs, by contrast, CO2 is liberated from haemoglobin in red blood cells, which, in turn, binds oxygen with a greater affinity.
Problem 10. Ion exchange capacity of a cation exchange resin
10. 1 The molecular formula of one unit of the catex polymer is C17H16O5S1, which corresponds to the molecular weight of 332. 369 g mol− 1. Mass percentage of an atom wx is

where ax and Ax are the number of atoms and the atomic weight of an atom X, respectively.
M is the molecular weight of one unit of the catex polymer. For sulfur (aS = 1,
AS = 32. 06 g mol− 1) and carbon (aC = 17, AC = 12. 011 g mol− 1), the mass percentage is
wS = 9. 65% and wC = 61. 43%, respectively.
10. 2 The theoretical ion exchange capacity is the amount of exchange groups in one unit of the catex polymer per mass of the unit, i. e.
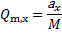
For -SO3H (one ion exchange group, aSO3H = 1) and -COOH (one ion exchange group
aCOOH = 1) we get Qm, SO3H = Qm, COOH = 3. 01 mmol g− 1.
10. 3 The total ion exchange capacity is a sum of individual strong and weak exchange capacities. For Qm, SO3H = Qm, COOH = 3. 01 mmol g− 1 we get Qm, total = 6. 02 mmol g− 1.
10. 4 The total ion exchange capacity in mmol cm− 3 of a swollen resin QV, total is
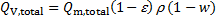
where ε and ρ are porosity and density, respectively, of a swollen resin and w is the mass ratio of water bound to the resin. For Qm, total = 6. 02 mmol g− 1, ε = 0. 48, ρ = 1. 28 g cm− 3, and w = 0. 45 we get QV, total = 6. 02 × (1 − 0. 48) × 1. 28 × (1 − 0. 45) = 2. 20 mmol cm− 3.
Problem 11. Weak and strong cation exchange resin
11. 1 At the beginning, all cation exchange sites are occupied with Na+ ions. Weak acetic acid exchanges all the weakly bound Na+ ions (weak cation exchange sites) and some of the strongly bound Na+ ions (strong cation exchange sites). The amount of Na+ in solution A is n1. When the resin is rinsed with a neutral solution of Mg2+ ions, all ions at the strong cation exchange sites are exchanged for Mg2+. Thus, solution B contains n2 moles of Na+ and n3 moles of H+.
The electrode potential is linearly proportional to the logarithm of concentration; i. e. for sodium ion selective electrode E = k + S log10[Na+]. Based on a two-point calibration, we get the following equations
− 0. 2283 = k + S log(0. 0100) and − 0. 3466 = k + S log(0. 00010)
Solving the system of equations, we get k = − 0. 1100 V and S = 0. 05915 V.
The amounts of Na+ ions in solutions A (VA = 1 000 cm3) and B (VB = 500 cm3) are
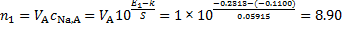 mmol
mmol
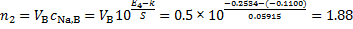 mmol
mmol
The alkalimetric titration is based on 1: 1 stoichiometry of the reaction of OH− (titration agent) and H+ (titrant). Then amount of H+ ions in solution B (Va is an aliquot of 100 cm3) is
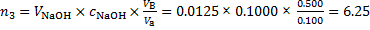 mmol
mmol
Ion exchange capacities of the strong and weak ion exchange resins (V0 = 4 cm3)
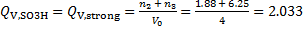 mmol cm− 3
mmol cm− 3
 mmol cm− 3
mmol cm− 3
11. 2 The total ion exchange capacity is
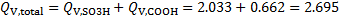 mmol cm− 3
mmol cm− 3
Problem 12. Uranyl extraction
12. 1 First, [HA]org is calculated:
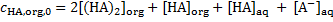
The concentration of UO2A2 is omitted as recommended in the introductory text.
From the definition of Kp, HA, KD, HA and Ka, HA, [HA]org can be obtained by solving the quadratic equation
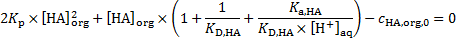
i. e.
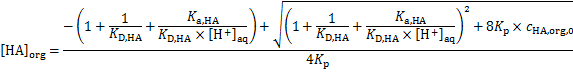
Considering that the proton concentration corresponds to the analytic concentration of HNO3, [H+]aq =  = 2. 00 × 10− 2 mol dm− 3, we get [HA]org = 3. 41 × 10− 3 mol dm− 3. Next, the uranyl ion distribution ratio,
= 2. 00 × 10− 2 mol dm− 3, we get [HA]org = 3. 41 × 10− 3 mol dm− 3. Next, the uranyl ion distribution ratio,  is expressed as:
is expressed as:
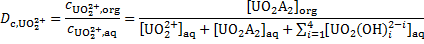
Using 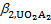 ,
,  ,
, 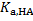 and bi for [UO2(OH)i]2–i complexes, can be expressed as
and bi for [UO2(OH)i]2–i complexes, can be expressed as
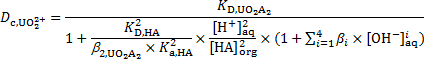
The concentration of hydroxyl ions is obtained from the concentration of protons,
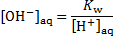
For [H+]aq = = 2. 00 × 10− 2 mol dm− 3, we get
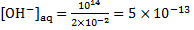 mol dm− 3
mol dm− 3
Casting this value, [HA]org = 3. 41 × 10− 3 mol dm− 3 and all the necessary constants into the expression for the distribution ratio, we obtain
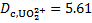
Then, the yield R defined as
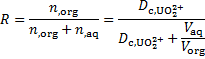
can be calculated, providing the final result of
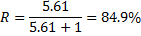
12. 2 For the conditions of [OH− ] = 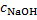 = 2. 00 × 10− 4 mol dm− 3, using the same calculation procedure, we get
= 2. 00 × 10− 4 mol dm− 3, using the same calculation procedure, we get
[HA]org = 1. 50 × 10− 5 mol dm− 3
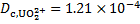
and the yield R = 0. 0121%.
Problem 13. Determination of active chlorine in commercial products
13. 1 (i) Cl2 + H2O → HClO ( A ) + HCl ( B )
(ii) NaClO + H2O → HClO ( A ) + NaOH ( C )
In alkaline aqueous solution, hypochlorite ion (ClO− ) will dominate.
13. 2

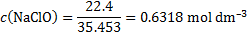
13. 3 ClO− + 2 I− + 2 H+ → I2 + H2O + Cl−
I2 + 2 S2O32− → 2 I− + S4O62−
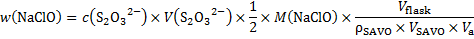
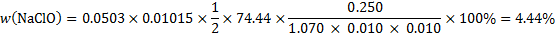
Problem 14. Chemical elements in fireworks
14. 1 An aqueous sample is introduced to a hot, non-luminous flame, where the tested compound is partially evaporated, atomized and free atoms are excited. During de-excitation, the energy difference between the atomic energy levels is emitted as a photon of an appropriate wavelength, characteristic of the particular chemical element. In this case, all three wavelengths are in the visible region of the spectrum and the corresponding colours for sodium, barium and lithium are yellow, lime green and red, respectively.
14. 2 The structure of a metal–EDTA complex is
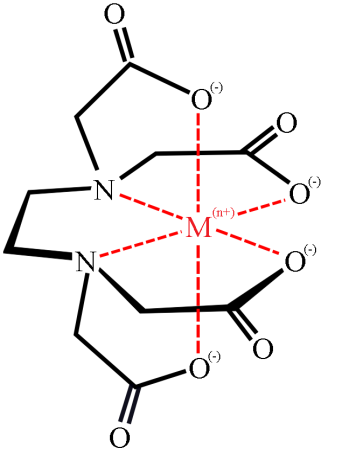
δ (HY3− ) = [HY3− ] / c(EDTA) = β 1 [H+] / (1 + β 1 [H+] + β 2 [H+]2 + β 3 [H+]3+ β 4 [H+]4)
δ (Y4− ) = [Y4− ] / c(EDTA) = 1 / (1 + β 1 [H+] + β 2 [H+]2 + β 3 [H+]3+ β 4 [H+]4),
where β 1 = 1 / Ka4, β 2 = 1 / (Ka4 Ka3), β 3 = 1 / (Ka4 Ka3 Ka2), β 4 = 1 / (Ka4 Ka3 Ka2 Ka1)
δ (HY3− ) = 1. 82 × 1010 × 10− 10 / (1 + 1. 82 × 1010 × 10− 10 + 2. 63 × 1016 × 10− 20 + 1. 23 × 1019 × 10− 30 + + 1. 23 × 1021 × 10− 40) = 0. 6453, i. e. 64. 53%
δ (Y4− ) = 1 / (1 + 1. 82 × 1010 × 10− 10 + 2. 63 × 1016 × 10− 20 + 1. 23 × 1019 × 10− 30 + 1. 23 × 1021 × 10− 40) = = 0. 3546, i. e. 35. 46%
δ (HY3− ) + δ (Y4− ) = 99. 99%, hence other forms are present at molar ratios lower than 0. 5%.
14. 3 Ammonium buffer is a mixture of ammonia and ammonium chloride. Ions of alkaline earth metals form weak complexes with EDTA (log KMY between 7. 7 and 10. 7) and are present only in alkaline media (pH > 9).
14. 4 Zn2+ + 4 CN− → [Zn(CN)4]2−
[Zn(CN)4]2− + 4 HCHO + 4 H2O → Zn2+ + 4 HOCH2CN + 4 OH−
14. 5 2, 3-Disulfanylpropan-1-ol is used for masking lead ions.
14. 6 Step i: Zinc is masked in the cyanide complex, lead and magnesium react with EDTA
n(Pb) + n(Mg) = n1(EDTA)
Step ii: EDTA released from its complex with lead ions reacts with magnesium standard solution
n(Pb) = nstd(Mg)
Step iii: Zinc released from cyanide complex reacts with EDTA
n(Zn) = n2(EDTA)
Masses of the elements in the sample (m1 = 1 g)
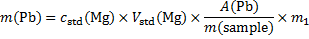

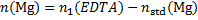
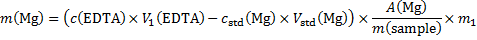
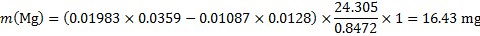

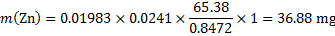
14. 7 Complexation equation: Ca2+ + Y4− → CaY2−
Final analytical concentrations after dilution are:
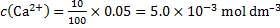
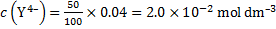
The coefficient for EDTA side reactions
α (EDTA) = (1 + β 1 [H+] + β 2 [H+]2 + β 3 [H+]3+ β 4 [H+]4)
(definitions of β i are in 14. 2); for pH = 6
α (EDTA) = (1 + 1. 82 × 1010 × 10− 6 + 2. 63 × 1016 × 10− 12 + 1. 23 × 1019 × 10− 18+ 1. 23 × 1021 × 10− 24) = = 4. 45 × 104
The final concentration of free Ca2+ ions in the solution is:
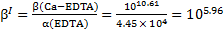
Based on the mass balances
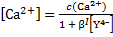
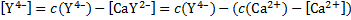
we get a quadratic equation with the following solution
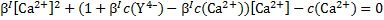
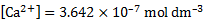
Problem 15. Colours of complexes
15. 1 Ground state: Excited state:
15. 2 The wavenumber of 20 300 cm− 1 corresponds to the wavelength of 493 nm which means the absorption of the blue-green light. The colour of the complex is the complementary one, i. e. orange-red.
15. 3 The complex absorbs visible light in the range from 493 to 575 nm, i. e. blue-green to
yellow-green. The complex is purple.
15. 4 Electron configurations:
15. 5 Equations:
(1) 2 CoCl2 + 3 F2 → 2 CoF3 + 2 Cl2
(2) CoF3 + 3 KF → K3[CoF6]
(3) 4 CoF3 + 2 H2O → 4 HF + 4 CoF2 + O2
15. 6 Equation:
(4) 4 CoCl2 + 4 NH4Cl + 20 NH3 + O2 → 4 [Co(NH3)6]Cl3 + 2 H2O
15. 7 The wavenumbers correspond to the wavelengths of 475 nm (blue light) and 340 nm (UV region). The second band has no effect on the observed colour and the complex is orange. Luteus means yellow in Latin (it refers to the yellow-orange colour of the complex).
15. 8 Due to their position in the spectrochemical series, fluoride ions (F− ) cause only small splitting, which leads to a high-spin configuration with four unpaired electrons. Ammonia molecules (NH3) cause greater splitting, which means that all the electrons in the t2g orbitals pair up and a low-spin configuration is formed
15. 9 Electron configurations:
15. 10 The wavenumbers correspond to the wavelengths of 877 nm (IR region) and 690 nm (red light). The first band has no effect on the observed colour and the complex is
blue-green.
Problem 16. Iron chemistry
16. 1 The requested potentials are calculated as follows, (1)–(3):
E°(FeO42− , H+/Fe2+) = (3 × 1. 90 + 1 × 0. 77) / 4 V = 1. 62 V (1)
E°(FeO42− , H+/Fe) = (3 × 1. 90 + 1 × 0. 77 + 2 × (− 0. 44)) / 6 V = 0. 93 V (2)
E°(Fe3+/Fe) = (1 × 0. 77 + 2 × (− 0. 44)) / 3 V = − 0. 04 V (3)
The corresponding Latimer diagram is depicted in Figure 1.

Figure 1. Latimer diagram for iron species in water (pH 0).
16. 2 The voltage equivalent is defined as a product of the formal oxidation state N and the standard redox potential E° (4) for the reduction of the particular species to the elemental state. The Frost diagram (Figure 2) then plots voltage equivalents versus oxidation state.
Voltage equivalent = N × E°(species/element) (4)
The individual voltage equivalents are calculated from the data above (5)–(8).
Voltage equivalent (Fe) = 0 × 0 V (def. ) = 0 V (5)
Voltage equivalent (Fe2+) = 2 × (− 0. 44) V = − 0. 88 V (6)
Voltage equivalent (Fe3+) = 3 × (− 0. 04) V = − 0. 12 V (7)
Voltage equivalent (FeO42− ) = 6 × 0. 93 V = 5. 58 V (8)
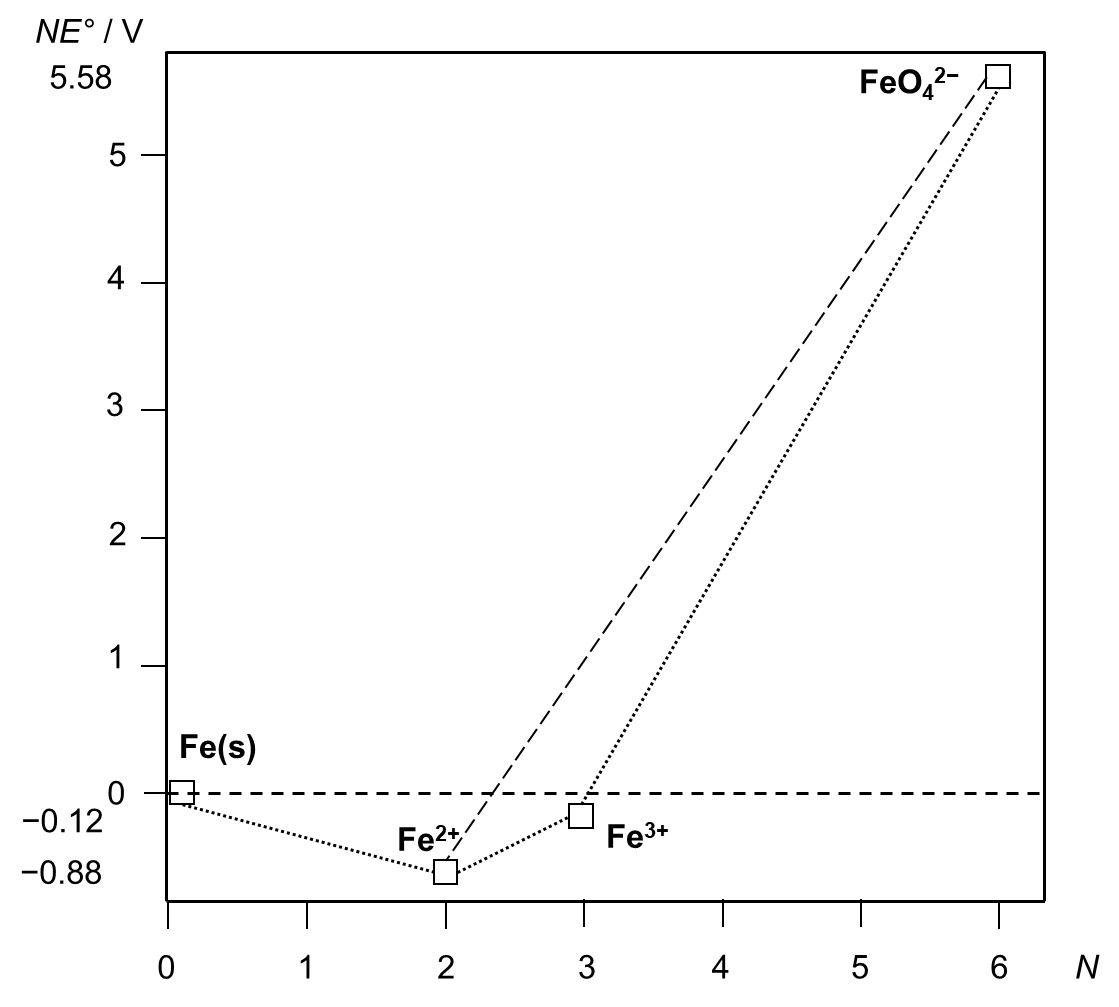
Figure 2. Frost diagram for iron species (pH 0).
Since the imaginary line connecting both the requested oxidation states (FeO42− and Fe2+, dashed line in Figure 2) lies above the Fe3+ point in the diagram, a synproportionation will be favoured, (9):
FeO42− + 3 Fe2+ + 8 H+ → 4 Fe3+ + 4 H2O (9)
16. 3
(a) The individual zone labels follow the successive uptake of electrons (from up to down) and hydroxide anions as ligands (from left to right). The identity of any zone can be checked by comparing the appropriate borderline definitions. The answers are displayed in Figure 3.
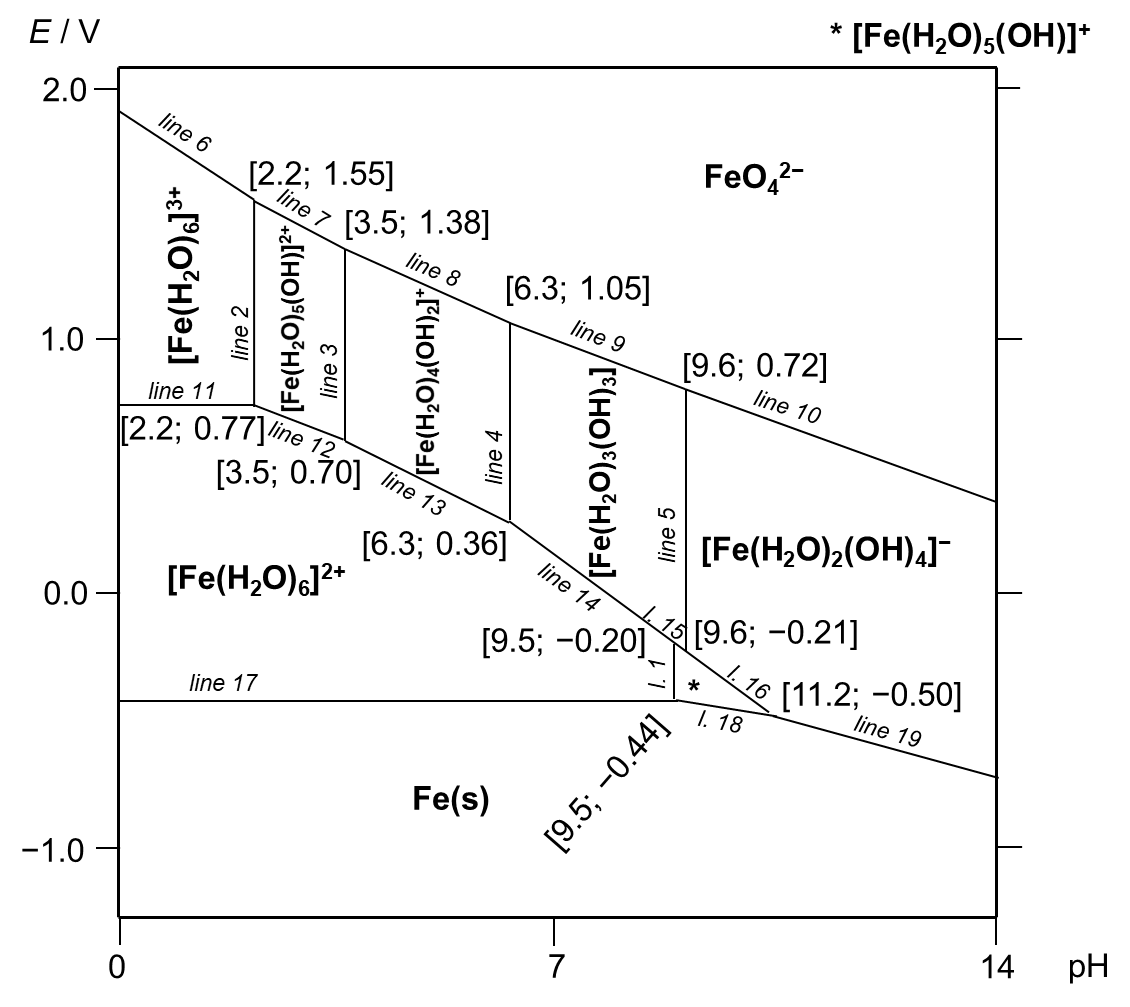
Figure 3. Pourbaix diagram for dissolved iron species and metallic iron.
It should be noted that this type of Pourbaix diagram would be valid only in highly diluted solutions of iron species. The increase in concentration will lead to the precipitation of insoluble (first ferric, then ferrous) oxides/hydroxides as well as the formation of polynuclear hydroxido complexes.
(b) Each borderline is drawn with the assumption that the activities of both participating species are equal. The equations which refer to lines 11 and 17, respectively, can be derived from the corresponding forms of the Nernst–Peterson equation (10) and (11) assuming the equilibrium conditions [Fe3+] = [Fe2+] and [Fe2+] = a(Fe, s). Because there is no term which depends on pH, the results are horizontal constant lines numerically equal to the standard redox potentials, see Figure 3.
line 11 (Fe3+/Fe2+): E = E° − 0. 059 × log([Fe2+] / [Fe3+]), thus E = 0. 77 (10)
line 17 (Fe2+/Fe): E = E° − (0. 059 / 2) × log(a(Fe, s) / [Fe2+]), thus E = − 0. 44 (11)
The conditions for lines 2 and 5 are [Fe3+] = [Fe(OH)2+] and [Fe(OH)3] = [Fe(OH)4− ], respectively, i. e. the expressions imply constant lines again, but in this case vertical ones, since there is no connection with the redox potential. The analytical expressions are represented by equations (12) and (13).
line 2 (Fe3+/[Fe(OH)]2+): pH = pKw − logβ 1, thus pH = 2. 2 (12)
line 5 ([Fe(OH)3]/[Fe(OH)4]− ): pH = pKw + logβ 3 − logβ 4, thus pH = 9. 6 (13)
(c) The analytic expression of line 6 (14) is also derived from the Nernst–Peterson equation under the assumption [FeO42− ] = [Fe3+].
line 6 (FeO42− /Fe3+): E = E° − (0. 059 / 3) × log{[Fe3+] / ([FeO42− ] × [H+]8)},
thus E = 1. 90 − 0. 157 × pH (14)
The first coordinate of the intersection of lines 2, 6 and 7 is obviously pH = 2. 2. The second coordinate can be calculated by substituting for pH = 2. 2 in equation (14), i. e. E = 1. 55.
Although it was not required to derive the expressions for all the lines, they are given in the following list for completeness, (15)–(28), and shown in Figure 3.
line 1 (Fe2+/[Fe(OH)]+): pH = 9. 5 (15)
line 3 ([Fe(OH)]2+/[Fe(OH)2]+): pH = 3. 5 (16)
line 4 ([Fe(OH)2]+/[Fe(OH)3]): pH = 6. 3 (17)
line 7 (FeO42− /[Fe(OH)]2+): E = 1. 86 − 0. 138 × pH (18)
line 8 (FeO42− /[Fe(OH)2]+): E = 1. 79 − 0. 118 × pH (19)
line 9 (FeO42− /[Fe(OH)3]): E = 1. 66 − 0. 098 × pH (20)
line 10 (FeO42− /[Fe(OH)4]− ): E = 1. 48 − 0. 079 × pH (21)
line 12 ([Fe(OH)]2+/Fe2+): E = 0. 90 − 0. 059 × pH (22)
line 13 ([Fe(OH)2]+/Fe2+): E = 1. 11 − 0. 118 × pH (23)
line 14 ([Fe(OH)3]/Fe2+): E = 1. 48 − 0. 177 × pH (24)
line 15 ([Fe(OH)3]/[Fe(OH)]+): E = 0. 92 − 0. 118 × pH (25)
line 16 ([Fe(OH)4]− /[Fe(OH)]+): E = 1. 48 − 0. 177 × pH (26)
line 18 ([Fe(OH)]+/Fe): E = − 0. 16 − 0. 030 × pH (27)
line 19 ([Fe(OH)4]− /Fe): E = 0. 38 − 0. 079 × pH (28)
(d) Ferrate ion can only be produced in a very basic solution by strong oxidizing agents (stronger than elemental oxygen under these conditions), e. g. hypochlorite (32). This will overcome line 10 and produce some ferrate ions.
2 [Fe(OH)4]− + 3 ClO− + 2 OH− → 2 FeO42− + 3 Cl− + 5 H2O (32)
Other possibilities are for example oxidation in a mixture of melted sodium nitrate with sodium hydroxide or analogous reactions in melts.
16. 4 From the viewpoint of " Hard and Soft Acid-Base" (HSAB) theory, Fe2+ is an intermediary hard, Fe3+ hard and imaginary " Fe6+" would be an extremely hard acid (the hardness correlates with the ionic radii and the surface charge density). Hard acids prefer hard bases and soft acids prefer soft bases. In aqueous solutions, H2O, OH− and O2− are available (although the oxide ion is not present in water at all, it can be at least formally " extracted" by e. g. a precipitation process). Therefore, the tendency to attract harder OH− ions is higher for Fe3+ than for Fe2+ which leads to a higher proton acidity of the hexaaquaferric ion and easier hydrolysis to the corresponding hydroxido species. For the Fe6+ centre, only a similarly extremely hard base is acceptable, i. e. O2− ion (although only formally, since there are covalent bonds between Fe(VI) and O atoms). Therefore, iron in the oxidation state +6 exists only in an anionic form.
16. 5 The answers are summarized in Table 1.
Table 1: Electronic and magnetic properties of selected iron species.
| Species | Conf. | Spin state | Magn. | LFSE | |
| [Fe(H2O)6]2+ | d6 | high- | t2g4eg2 | para- | − 0. 4Δ o + P |
| [Fe(CN)6]4− | d6 | low- | t2g6eg0 | dia- | − 2. 4Δ o + 3P |
| [Fe(H2O)6]3+ | d5 | high- | t2g3eg2 | para- | 0 |
| [Fe(H2O)5OH]2+ | d5 | high- | (t2g3eg2)* | para- | 0 |
| [Fe(CN)6]3− | d5 | low- | t2g5eg0 | para- | − 2. 0Δ o + 2P |
Remark: * not Oh symmetry.
16. 6 a) FeCl3, b) Fe4[Fe(CN)6]3.
Problem 17. Cyanido- and fluorido-complexes of manganese
17. 1 (1) 2 Mn + 12 NaCN + 2 H2O → 2 Na5[Mn(CN)6] + H2 + 2 NaOH
17. 2 Diagram – low-spin configuration d6:
17. 3 Diagram – high-spin configuration d5:
The complex has five unpaired electrons.
17. 4 Diagram – low-spin configuration d5:
17. 5 Equations:
(2) 4 Mn2+ + O2 + 24 CN− + 2 H2O → 4 [Mn(CN)6]3− + 4 OH−
(3) 2 [Mn(CN)6]4− + H2O2 → 2 [Mn(CN)6]3− + 2 OH−
(4) 3 MnCl2 + HNO3 + 3 H3PO4 → 3 MnPO4↓ + NO + 6 HCl + 2 H2O
(5) MnPO4 + 6 KCN → K3[Mn(CN)6] + K3PO4
17. 6 Diagram – low-spin configuration d4:
17. 7 (6) 4 MnO2 + 12 KHF2 → 4 K3[MnF6] + O2 + 6 H2O
17. 8 Diagram – high-spin configuration d4:
17. 9 Sharing 1 bridging F atom between 2 neighbouring octahedral units corresponds to the stoichiometry [MnF5]2− :
17. 10 The stoichiometry [MnF4]− could be achieved in a chain structure having 2 bridging F atoms between 2 neighbouring octahedral units:
However, the structure is a 2D-anionic layer, so it is necessary to extend the structure to 2 dimensions – to have 4 bridging F atoms for each octahedral unit:
17. 11 Diagram – configuration d3:
17. 12 Since the magnitude of splitting in tetrahedral crystal field is about a half of octahedral (exactly Δ tet = 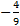 Δ oct; the negative sign refers to the inverse order of split d-orbitals with respect to the octahedral crystal field), it is always lower than electron pairing energy (Δ tet < P) which leads to high-spin configurations in tetrahedral complexes.
Δ oct; the negative sign refers to the inverse order of split d-orbitals with respect to the octahedral crystal field), it is always lower than electron pairing energy (Δ tet < P) which leads to high-spin configurations in tetrahedral complexes.
Diagram – high-spin configuration d5:
17. 13 (7) 2 KMnO4 + 3 H2O2 + 2 KHF2 + 8 HF → 2 K2[MnF6] + 3 O2 + 8 H2O
17. 14 Manganese(IV) complexes have a configuration of d3. Since these three electrons occupy the t2g level only, low- and high-spin configurations cannot form regardless of the magnitude of the crystal field splitting.
17. 15 (8) K2[MnF6] + 2 SbF5 → 2 K[SbF6] + MnF2 + F2
Problem 18. The fox and the stork
Large stones
18. 1 Each layer consists of one sphere only: n = 50 / (2 × 5) = 5
18. 2 The volume of 5 spheres: 
The volume of cylinder: 
The fraction of volume: f = 2 618 / 3 927 = 0. 667, i. e. 66. 7 %
18. 3 The free volume: Vfree = 3 927 − 2 618 = 1 309 cm3
|
|
|
© helpiks.su При использовании или копировании материалов прямая ссылка на сайт обязательна.
|